Aldehydes and Ketones
Hydrates, Hemiacetals, and Acetals
Last updated: May 28th, 2026 |
Hydrates, Hemiacetals, and Acetals – Their Formation From Aldehydes/Ketones, And Mechanisms That Are As Easy As P-A-D-P-E-A-D
Hydrates, hemiacetals and acetals are the products of addition reactions of oxygen-based nucleophiles (water and alcohols) to aldehydes and ketones
- A hydrate contains a carbon with two single bonds to OH.
- A hemiacetal contains a carbon with a single bond to OH and a single bond to OR (where R is a carbon group)
- An acetal (sometimes called a ketal if originating from a ketone) contains a carbon with two single bonds to OR groups.
In this article and in the quizzes at the bottom, you will learn to identify these functional groups and how they are made (and removed)
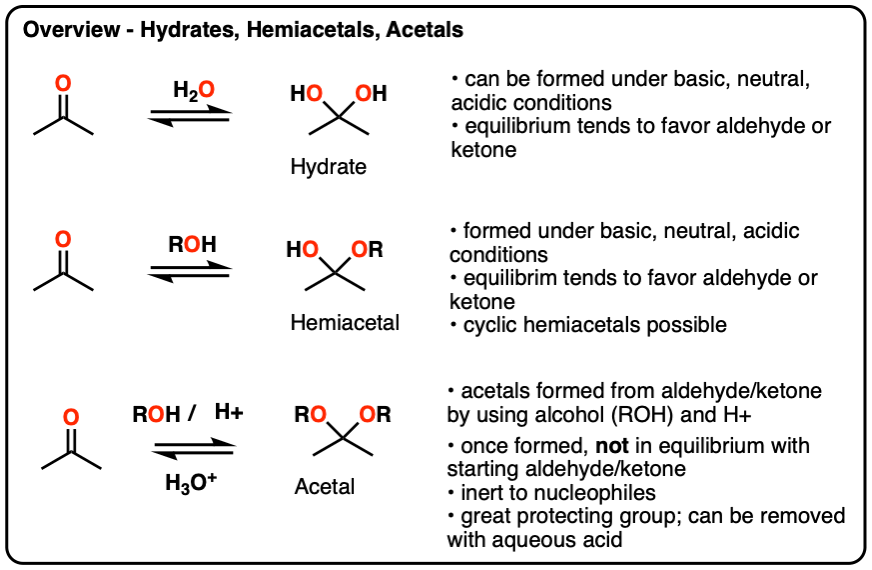
Hydrates and hemiacetals are in equilibrium with their respective aldehydes and ketones.
Acetals are formed through treatment of an aldehyde or ketone with an alcohol in the presence of (anhydrous) acid.
Unlike hydrates and hemiacetals, acetals are “locked”, and are not in equilibrium with their corresponding aldehyde/ketone. For this reason, acetals are useful protecting groups for aldehydes/ketones.
Acetals can be converted back to the starting aldehyde/ketone with aqueous acid (H3O+).
Thiols can be used in place of alcohols to make thioacetals. The most common reaction of thioacetals is their reduction with Raney nickel to give the corresponding alkanes.
Table of Contents
- Hydrates – Their Formation and Mechanism
- Hemiacetals – Formation and Mechanism
- What about Intramolecular Hemiacetal Formation?
- Formation of Acetals From Hemiacetals
- Formation of Acetals From Aldehydes and Ketones -As Easy As PADPEAD
- Cyclic Acetals
- Reactions of Acetals
- Acetals as Protecting Groups
- Thioacetals (and their Reactions)
- Summary
- Notes
- Quiz Yourself!
- (Advanced) References and Further Reading
1. Hydrates
Hydrates, hemiacetals, and acetals. What are they, how are they formed, what are their properties, and most importantly – why should you care?
Let’s start with hydrates, which are the simplest . They happen to be the one example of the three you should care the least about, but hey – they’re illustrative!
A hydrate is formed when aldehydes or ketones react with water (H2O). Hydrates contain a carbon single-bonded to two OH (hydroxyl) groups [ C(OH)2]. [Note 1 – Hydrates are also known as “gem-diols”]
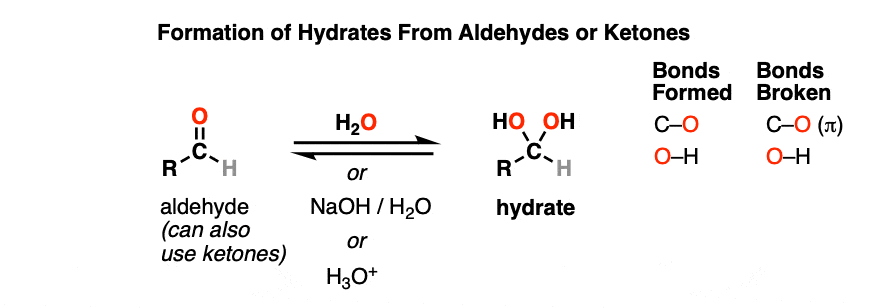
Note the carbon attached to two OH groups. In the reaction between the aldehyde and water, a new C-OH bond has formed and a C-O pi bond has been broken. Another way of saying this is that net addition of H2O has occurred across the C=O bond.
Hydrate formation is generally reversible, and in any solution containing a hydrate there will be an equilibrium between the starting aldehyde or ketone and the hydrate product. Equilibrium generally (but not always, Note 2) favors the starting aldehyde or ketone.
Hydrates are relatively stable in solution but are difficult to isolate in their pure form. Once solvent H2O is removed, equilibrium tends to favor reversion back to the starting aldehyde/ketone. [Note 3 – This is classic “Le Chatelier”]
Hydrates can be formed under neutral (H2O), basic (NaOH/H2O) or even acidic (H3O+) conditions.
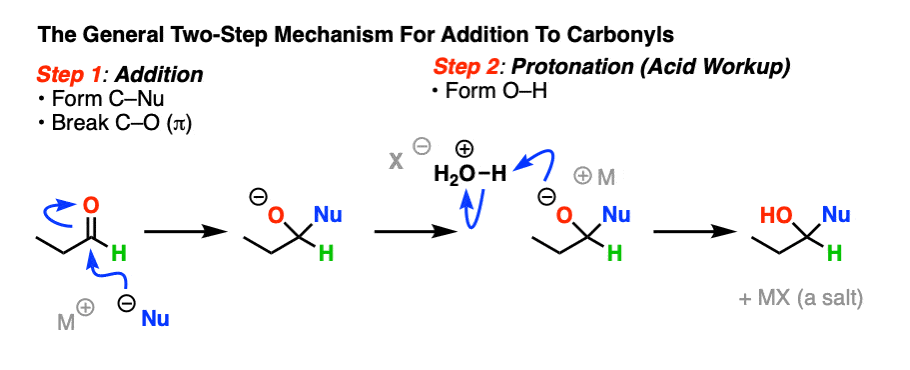
The simplest mechanism for hydrate formation is under basic conditions (e.g. NaOH / H2O). It consists of
1) addition of the nucleophile (HO- ) to the carbonyl carbon [forming C-OH, breaking C-O (pi)] followed by
2) protonation of the oxygen (form O-H).
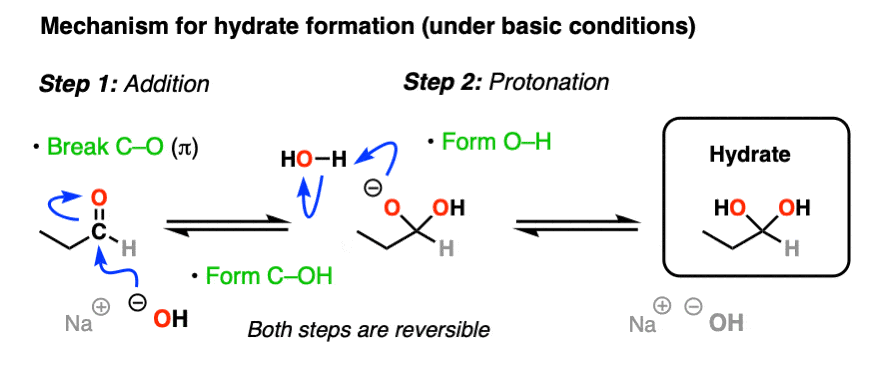
If this mechanism looks familiar, it should! Astute readers may note that this is exactly the same as the classic “2 step” mechanism we recently explored for aldehydes/ketones shared by at least 7 other prominent reactions (reduction, Grignard addition, cyanohydrin formation, etc) – addition followed by protonation.
Classic 2-step mechanism (hover) or click link.
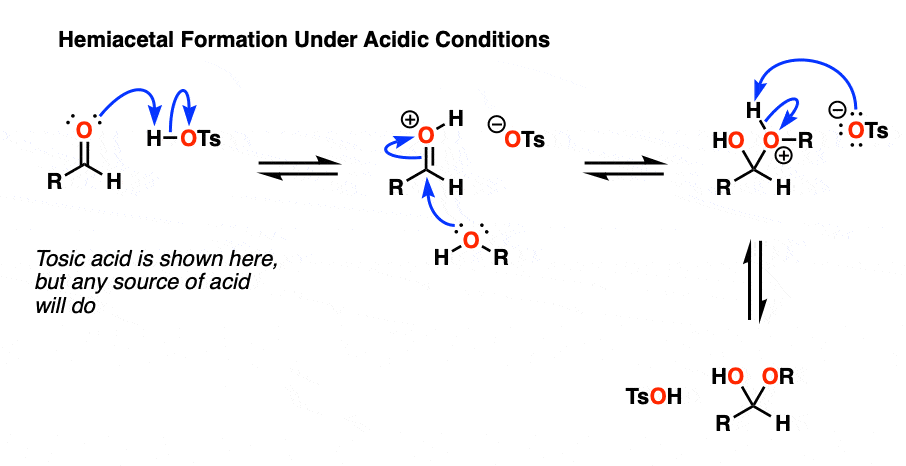
Under acidic conditions the mechanism is slightly longer since we have to account for an extra proton transfer step. One way to draw it is to start with 1) protonation of the aldehyde/ketone oxygen with acid, followed by 2) addition of H2O, and then 3) deprotonation of H2O with mild base.
Mechanism for acidic hydrate formation (hover) or click this link.
2. Hemiacetals
When aldehydes or ketones react with alcohols they form hemiacetals. A hemiacetal resembles a hydrate except that one of the OH groups has been replaced by OR.
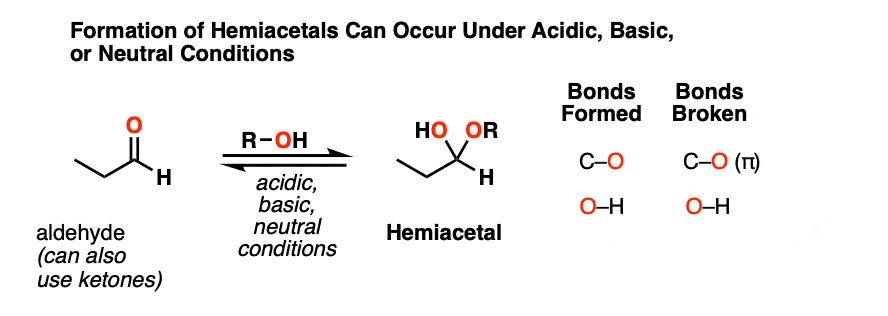
This is the net addition of an alcohol (ROH) across the carbonyl group. A C-O bond forms, a C-O pi bond breaks, and a new O-H bond forms.
As with hydrate formation, hemiacetal formation can happen under basic, neutral or acidic conditions.
Basic conditions [RO– , ROH] provide another example of the classic “two-step” addition-protonation mechanism so common to these functional groups (addition of RO(-) followed by protonation).
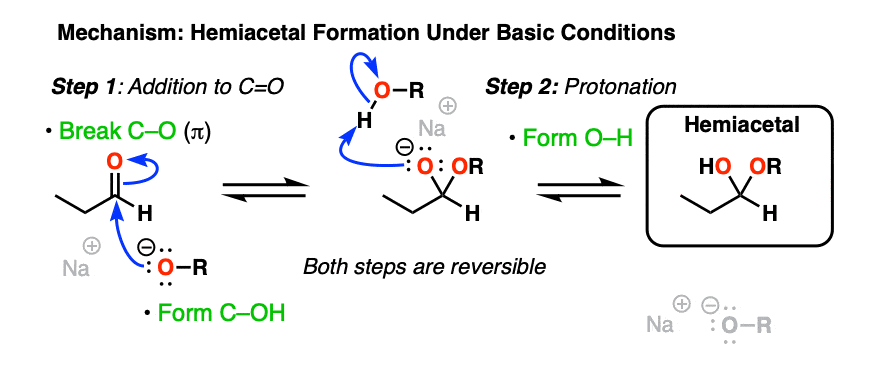
As with hydrates, the mechanism under neutral or acidic conditions requires an extra proton transfer step.
One way to draw the mechanism for formation under acidic conditions is 1) protonation of the aldehyde / ketone oxygen with acid, followed by 2) addition of neutral alcohol, and then 3) deprotonation of the oxygen with weak base.
Mechanism for acid-catalyzed hemiacetal formation (hover) or click link.
Equilibrium generally favors the starting aldehyde/ketone. Likewise, isolation of pure acyclic hemiacetals is often difficult since removal of solvent generally leads to loss of the alcohol and restoration of the starting aldehyde / ketone. On the other hand, cyclic hemiacetals tend to be considerably more stable. [See post: Ring-Chain Tautomerism]
3. Intramolecular Hemiacetal Formation
If an alcohol and an aldehyde /ketone are present on the same molecule, there is always the possibility that they will combine to form a cyclic hemiacetal.
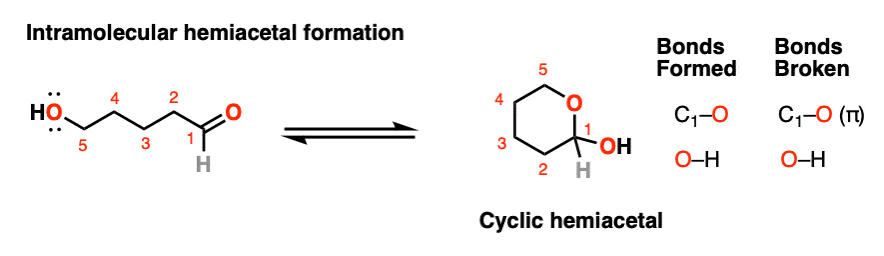
Note that the reaction is fundamentally the same as in the intermolecular case. The mechanism for the formation of a cyclic hemiacetal is no different from that of an acyclic acetal.
We’re still forming C-O, breaking C-O (pi), and forming O-H. That’s it. All that’s different is that the OH and the carbonyl are connected through a carbon chain. It takes some getting used to, but as I often tell my students, it’s conceptually no more different than the loop that results when you put a “nucleophilic” belt-buckle pin through an “electrophilic” belt notch.
Mechanism for intramolecular hemiacetal formation (hover) or click link.
If they can combine to form a 5- or 6-membered ring (i.e. reasonably free of ring strain) equilibrium will generally favor the cyclic form. Most simple carbohydrates exist predominantly as cyclic hemiacetals. For example, in water at 25° C, a solution of glucose contains less than 1% of the open-chain form at any given time! Likewise, fructose and ribose (and many other sugars, besides) are usually drawn as cyclic hemiacetals.
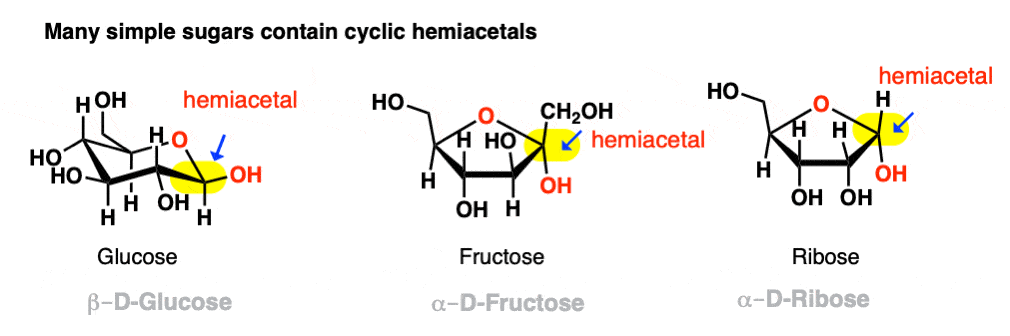
Even if equilibrium strongly favors the cyclic hemiacetal (often the case with 5- and 6-membered rings) there will always be a small concentration of the hemiacetal in its linear (“acyclic”) form with the parent aldehyde/ketone. This behavior goes by the fancy name of ring-chain tautomerism .
If you continue on to the chapter on carbohydrates later in Org 2, you will learn that this is also responsible for the property of mutarotation in sugars whereby the optical rotation of a solution of certain forms of glucose can change over time.
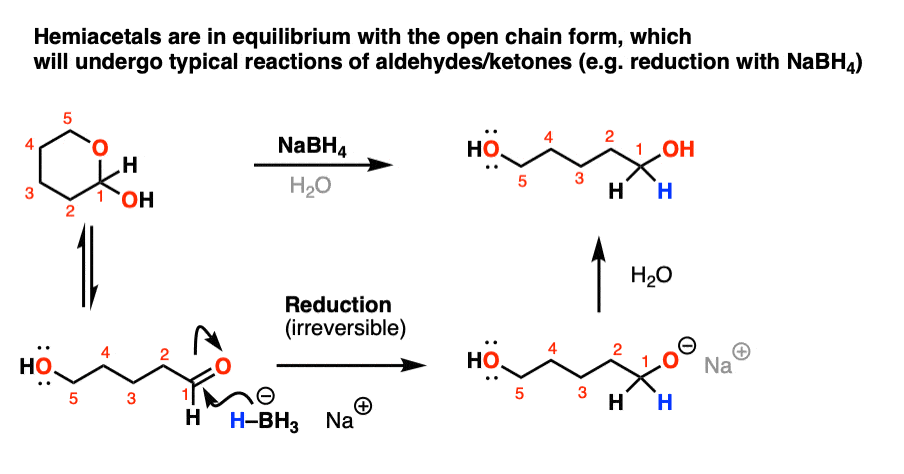
The reversibility of hemiacetal formation in solution also means that despite their appearances, cyclic hemiacetals have the same reactivity as the parent aldehyde/ketone since the two forms are interchanging rapidly in solution.
For example, if a cyclic hemiacetal is treated with the reducing agent NaBH4 it will be quickly be reduced irreversibly to the acyclic alcohol.
Reduction of cyclic hemiacetals (hover) or click link.
4. Formation of Acetals From Hemiacetals
A carbon that is single-bonded to two OR groups is generally referred to as an acetal. (The term “ketal” also gets used to describe acetals originating from ketones, but we’ll stick with “acetal” here). [Note 4]
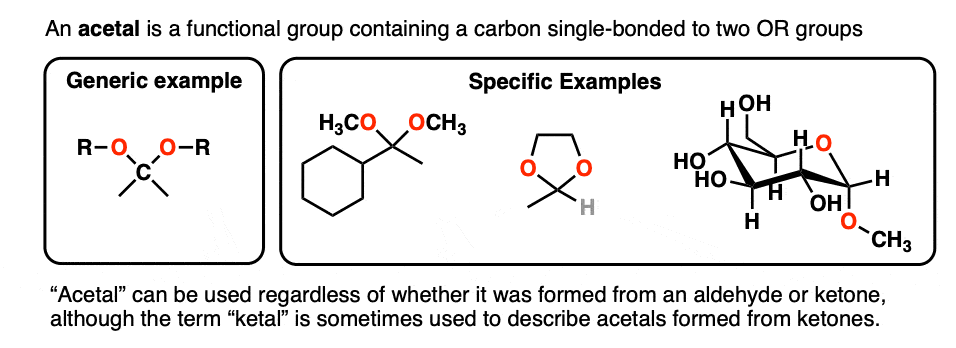
So how are they made?
If hydrates and hemiacetals are made through the net addition of H2O and ROH across a C-O pi bond, respectively, you might note a dilemma here.
There’s no C-O pi bond to add our second equivalent of ROH across!
If you just dissolve a hemiacetal in solution in the presence of excess alcohol, pretty much no acetal will be formed.
The hemiacetal needs a kick in the pants!
Now, if we add (anhydrous) acid, we’ll end up protonating the OH group to give H2O+ . [Note 5 – why doesn’t OR get protonated here?] As we’ve seen many times before, the conjugate acid is a better leaving group. This sets up the second-most important mechanism of carbonyls, elimination of a leaving group to form a new C-O pi bond where the O bears a positive charge. [Note 6 ].
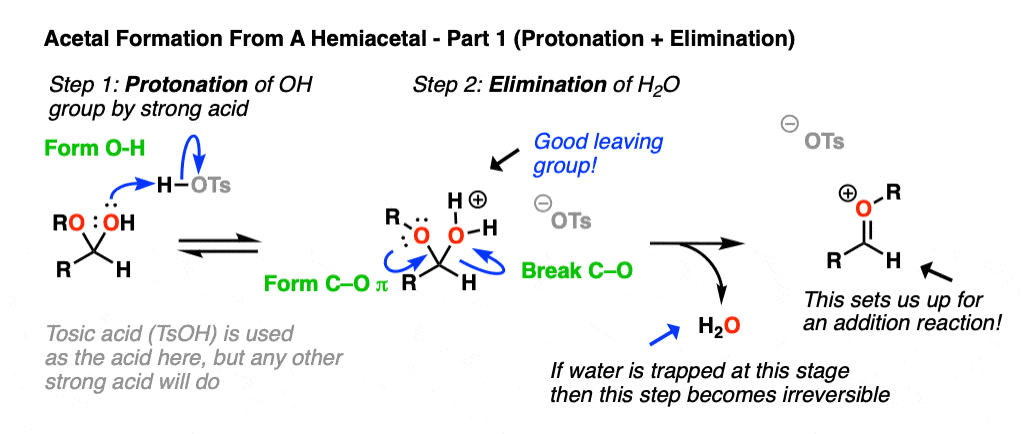
Now we have a C-O pi bond that we can add our second equivalent of ROH across.
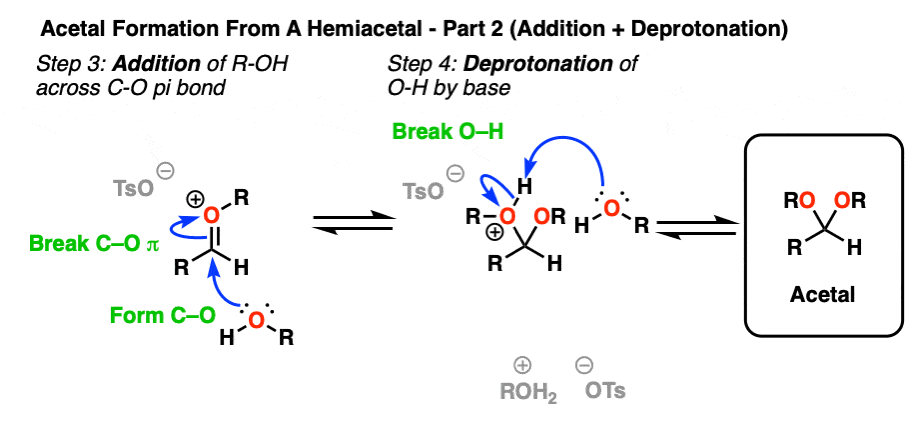
Addition of ROH, followed by deprotonation, gives us our neutral acetal.
5. Formation of Acetals From Aldehydes and Ketones
It’s good to know how acetals are formed from hemiacetals. But most of the time, we’ll be seeing examples of aldehydes and ketones being converted into acetals, like this.
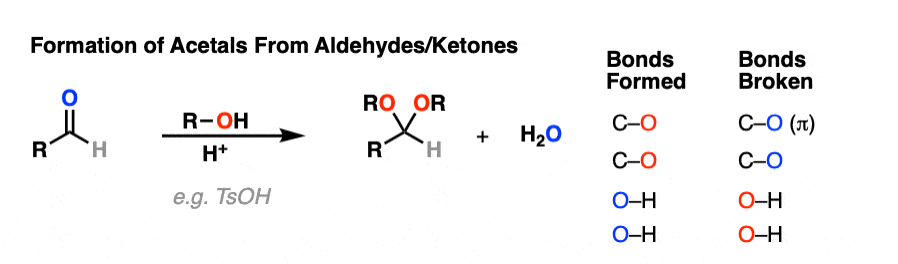
So how does that work?
Turns out that the whole mechanism for conversion of an aldehyde/ketone to an acetal is nothing more than
- Conversion of an aldehyde/ketone to a hemiacetal (under acidic conditions),
followed by - Conversion of a hemiacetal to an acetal
We’ve already covered both of these steps! So all we’re going to do here is put everything together into one extended sequence.
In the chemistry of carbonyls (like aldehydes and ketones) there’s a lot of repetition of just a handful of mechanistic steps. So you might find it helpful to abbreviate some of these steps as P (protonation) D (deprotonation) A (addition) and E for (elimination) because this will help you remember the mechanisms more easily.
Going from an acetal to a hemiacetal is P A D. The first step is protonation of the aldehyde, followed by addition of the alcohol nucleophile, and deprotonation of the oxygen to give the neutral hemiacetal.
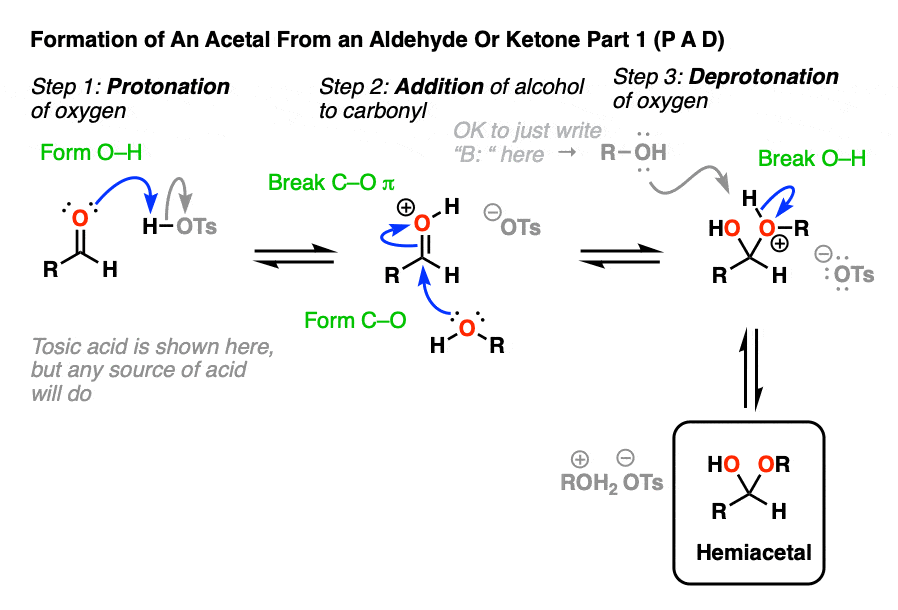
Note that I drew in tosic acid (TsOH) as the acid catalyst here, but more often than not, a simple H+ will be sufficient here.
Going from the hemiacetal to the acetal is P E A D. The OH is protonated, followed by elimination of water, addition of the second equivalent of the alcohol, and then deprotonation to give the neutral hemiacetal.
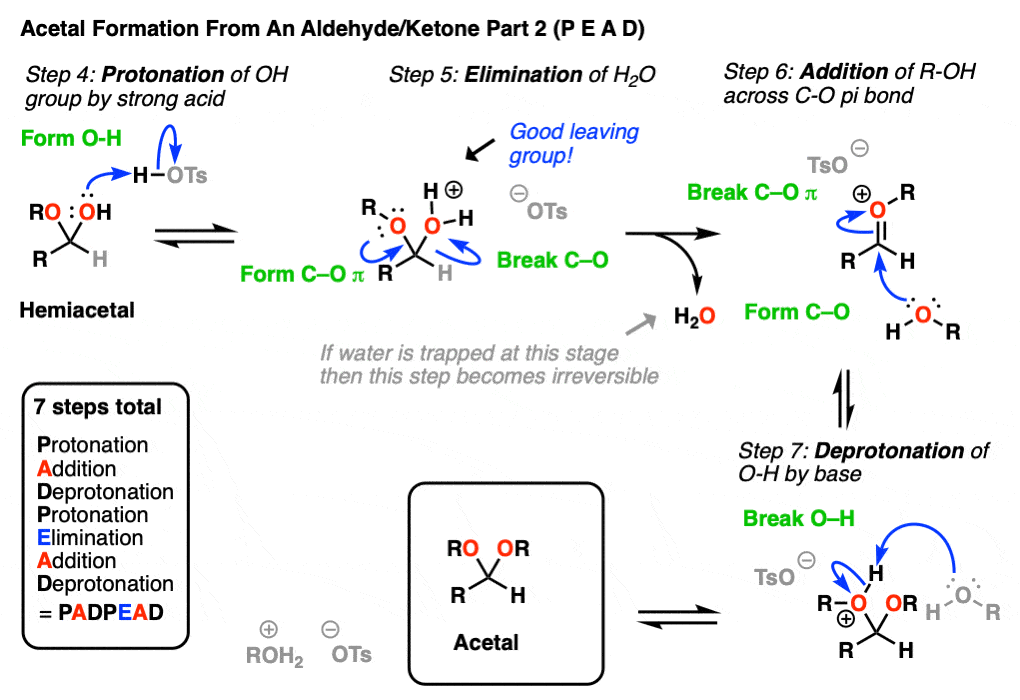
So putting everything together we get P A D P E A D, which is a pretty easy mnemonic to remember.
[One note – one common shorthand term for Protonation-Deprotonation (or its converse D-P) is “proton transfer”. In upper level courses it’s not uncommon to omit drawing out the protonation-deprotonation steps and just wave The Magic Wand Of Proton Transfer. because, let’s face it, life is short]
One important thing to note about acetal formation is that every step is potentially reversible. This presents us with a small dilemma. If we want to make the acetal, for example, how can we drive the equilibrium towards the desired product?
The first consideration for driving the equilibrium forward is to use a large excess of ROH relative to the molar equivalents of H2O that are formed. The second consideration, which can be used in combination with the above, is to employ some kind of a drying agent (desiccant) that will react with any H2O is formed, or to sequester H2O in a Dean-Stark trap.
From Organic Syntheses, here’s a link to a classic procedure for making the diethyl acetal of acetaldehyde. CaCl2 serves as a drying agent as well as a Lewis acid for acetal formation.
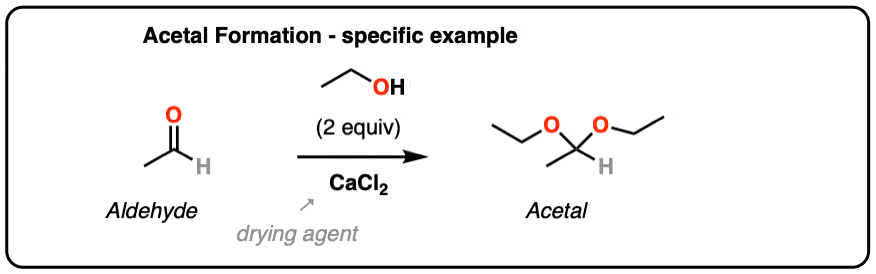
[H. Adkins and B. H. Nissen, Org Syn Coll. 1. Page 1. ]
6. Cyclic Acetals
There’s another wrinkle in acetal formation that deserves mention, especially as it seems to make its way on to a lot of exams.
In all of our examples up to this point, we’ve used “ROH” where it’s assumed that R is a short-chain alkyl group of some sort (e.g. CH3OH, CH3CH2OH).
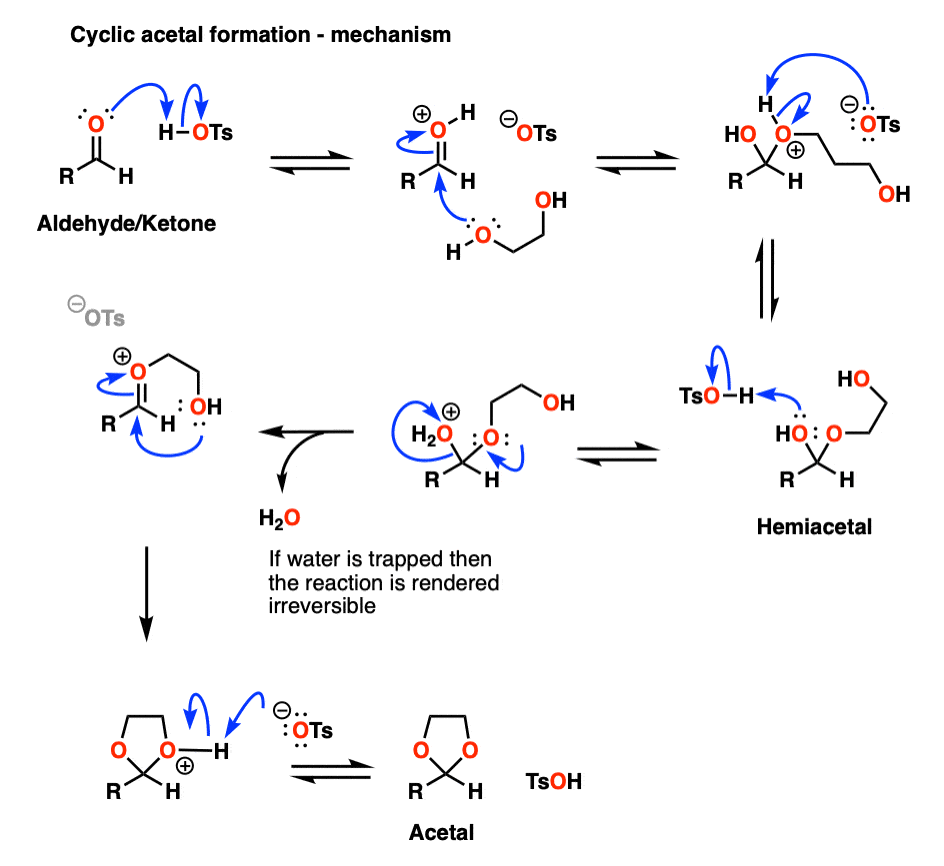
It turns out that instead of using 2 equivalents of a short-chain alcohol ROH, you can use 1 equivalent of a di-ol such as ethylene glycol. This results in a cyclic acetal. [Note 7]
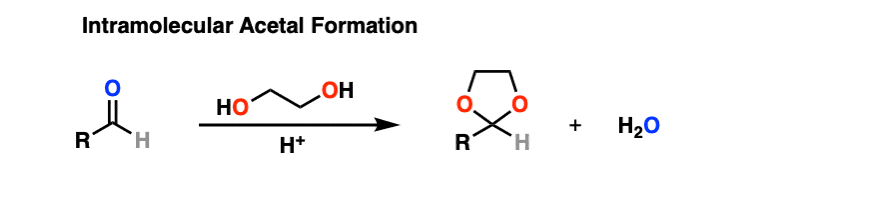
So how does this work?
It’s really no different than the intermolecular case. The bonds that form and break are the same. The mechanism is the same. It’s still P A D P E A D. That said, here’s a link to the image if you need to see it for yourself.
Mechanism for formation of a cyclic acetal (hover) or click link.
BTW, there is a second possibility for making a cyclic acetal that is great exam question fodder. It requires a molecule that contains a ketone and two hydroxyl groups. Adding acid makes it wrap up into a tidy little bow. See Note 8 for details.
7. Reactions Of Acetals
So what kinds of reactions do acetals undergo? As it turns out, not much.
We saw previously that hemiacetals, being in equilibrium with the parent aldehyde/ketone, pretty much undergo the same reactions as aldehydes and ketones.
Not so with acetals.
Once formed, acetals are “locked” into place and are not in equilibrium with their parent aldehyde/ketone. They’re solid as a rock under basic and neutral conditions. NaBH4, LiAlH4, Grignard reagents, organolithiums, PCC, you name it. Acetals are impregnable.
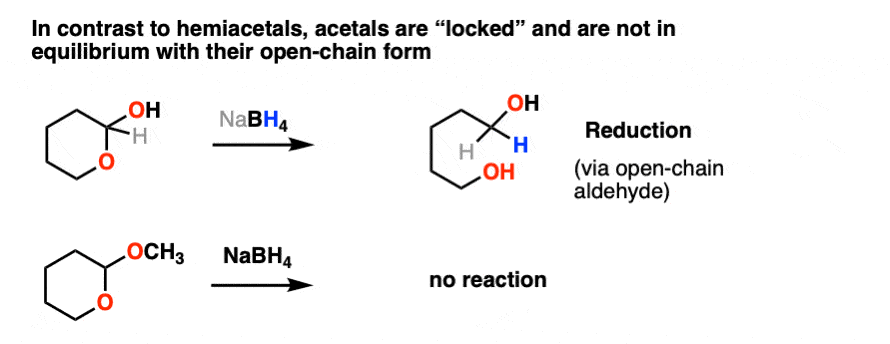
Like ethers – another contender for the “Most Boring Functional Group” award – acetals pretty much undergo only one reaction. When treated with aqueous acid, they can be hydrolyzed back to the starting aldehyde/ketone.
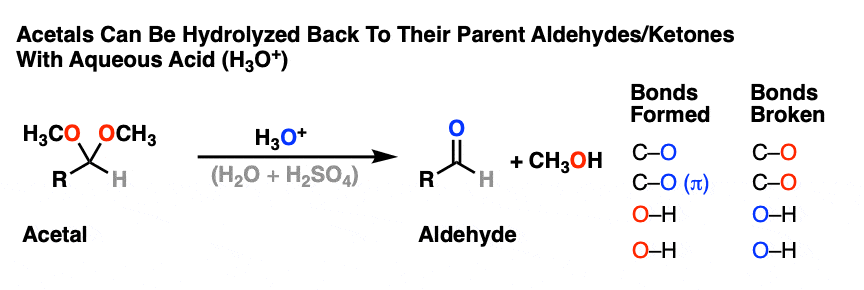
That’s it. You’ve seen all the reactions of acetals you need to see.
So how does this work? Let me spoil the suspense and tell you that it is some combination of P, D, E, and A.
[Actually if you know that 1) aldehyde → acetal is PADPEAD, and 2) P and D are inverse reactions, as are A and E, then you can figure out in advance what the mechanism for acetal → aldehyde is. Note 9. ]
OK. We start with an acetal and add aqueous acid. The first step is Protonation. This gives us the good leaving group ROH, which is displaced by an Elimination to give a new C-O pi bond. The C-O pi bond undergoes Addition of water, and the oxygen is converted to its neutral form through Deprotonation. Then, Protonation of the other OR group gives us ROH, which undergoes Elimination to give the protonated aldehyde. Deprotonation gives us the neutral aldehyde.
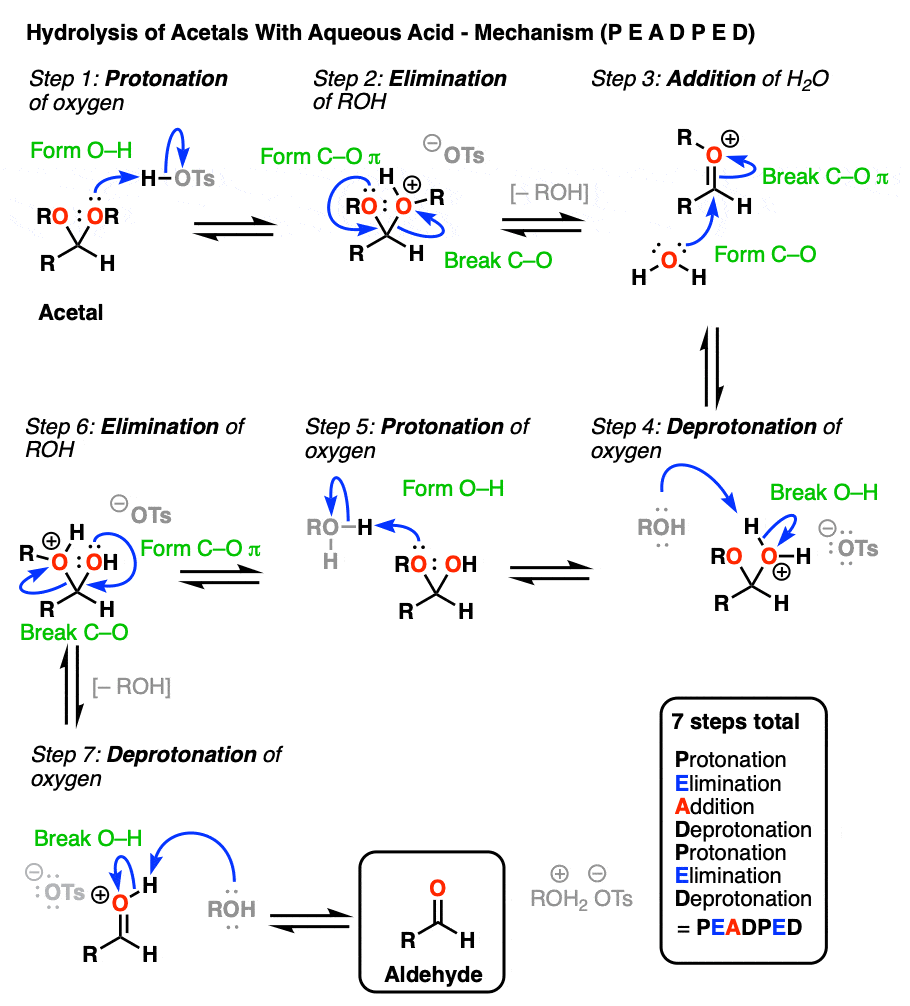
Although an aldehyde is depicted here, it also works for ketones.
To get the reaction to go to completion, it’s necessary to use a large excess of water, which drives the equilibrium towards the final product.
8. Acetals As Protecting Groups
There is a hidden benefit of being able to convert a reactive functional group (such as alcohols, aldehydes and ketones) to a Boring Functional Group (such as an ether or acetal) and back again.
As we saw with alcohols, it provides us with a way of putting the chemical equivalent of “painter’s tape” over a reactive functional group while we fiddle around with a different functional group on the other side of the molecule, content in the knowledge that it won’t interfere with our machinations.
For example, trying to make a Grignard reagent on a molecule that has an aldehyde or ketone is a no-go. As soon as the Grignard is formed, it will react with the aldehyde or ketone, giving us a big “BFM” to write in our lab notebook.

However, if we first protect the ketone as an acetal, then convert it into a Grignard reagent, we can Grignard it up as much as we want without fear that the ketone will be destroyed. After we’re finished Grignarding, it’s a simple matter to then treat the resulting product with aqueous acid, liberating the ketone, unscathed.
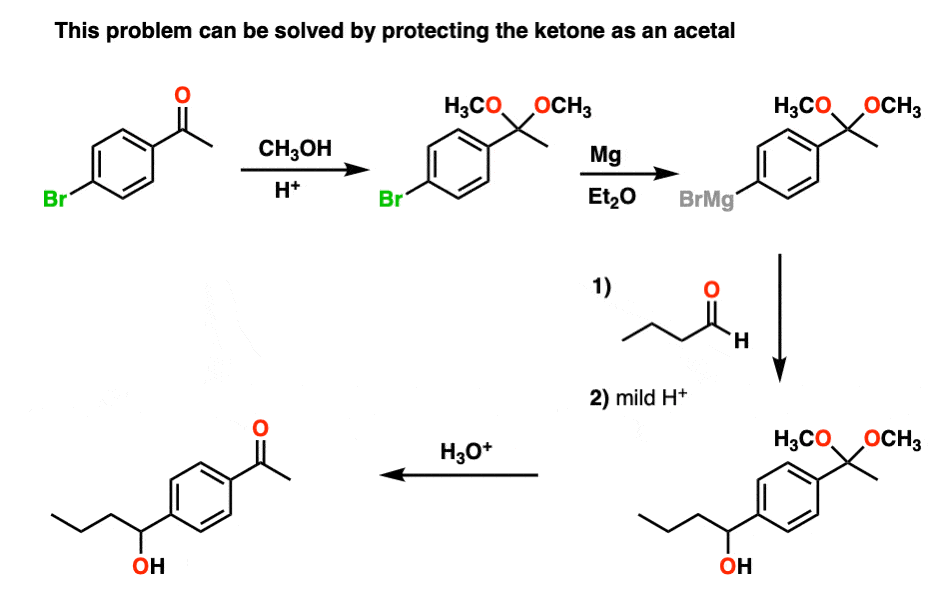
By the way, you can also look at it the other way around. If you make a diol (e.g. from dihydroxylation of an alkene), you can protect the diol as an acetal by treating with an aldehyde or ketone.
9. Thioacetals and Their Reactions
As is often the case, many reactions of OH groups translate pretty well when you move down the periodic table to SH, only things get a lot smellier.
By treating an aldehyde or ketone with 1,2-ethanedithiol (the di-thiol equivalent of ethylene glycol) and a Lewis acid like BF3 (plain old H+ doesn’t work as well when sulfur is involved) one can make a thioacetal.
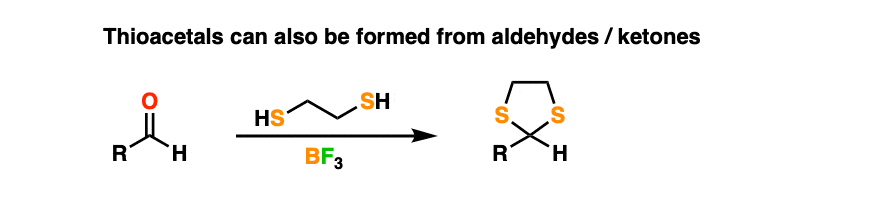
Thioacetals are perfectly serviceable protecting groups. However, they have one interesting feature that ordinary acetals lack.
By treating a thioacetal with Raney Nickel, one can effectively “delete” the thioacetal, replacing the C-S bonds with C-H bonds (Raney nickel is a finely divided nickel-aluminum alloy that contains adsorbed hydrogen).
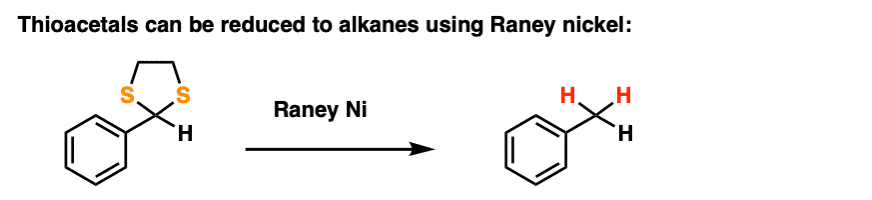
Along with the Wolff-Kishner and Clemmensen, it’s a nifty trick for getting rid of troublesome carbonyl groups and converting them to alkanes, a key component of the great Friedel-Crafts Workaround, by the way.
10. Conclusion
So what have we learned?
- Water and alcohols can add to aldehydes and ketones to give hydrates and hemiacetals. The equilibrium generally favors the aldehydes/ketones but cyclic hemiacetals are pretty stable.
- Still, cyclic hemiacetals behave like aldehydes/ketones as far as reactivity is concerned, since they’re in equilibrium
- Treating a hemiacetal with acid and an alcohol will convert it to an acetal
- Treating an aldehyde or ketone with an alcohol (or a diol) plus acid will convert it to an acetal, via P A D P E A D.
- Acetals only undergo one significant reaction – hydrolysis with aqueous acid, which occurs via P E A D P E D.
- Acetals are useful protecting groups for aldehydes/ketones
- Thioacetals are also useful, but mostly because they allow conversion to an alkane via Raney Ni.
Notes
This post was extensively re-written in Feb 2022, replacing an older post called “On Acetals and Hemiacetals”
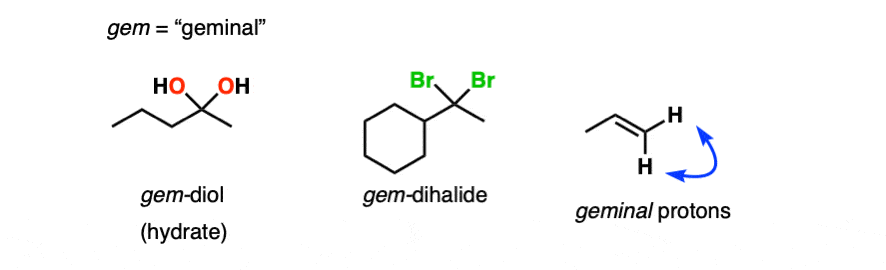
Note 1 – Another term for hydrates is “geminal diols” (gem-diols). Geminal refers to two groups bonded to the same carbons. We encountered gem-dihalides in the addition of HCl and HBr to alkynes, for example.
Note 2. Electron-withdrawing groups stabilize the hydrate form. The hydrate of chloraldehyde [ Cl3CCHO ], for example, is a colorless solid.
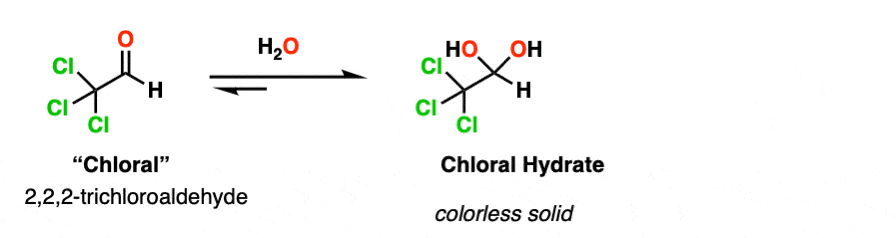
Chloral hydrate was the earliest hydrate to be isolated (in 1832) and saw early use as an anaesthetic. It is probably the most famous hydrate of all, playing a prominent role in numerous spy and detective films as a “Mickey Finn” or “knockout drug”.
From The Living Daylights, 1987
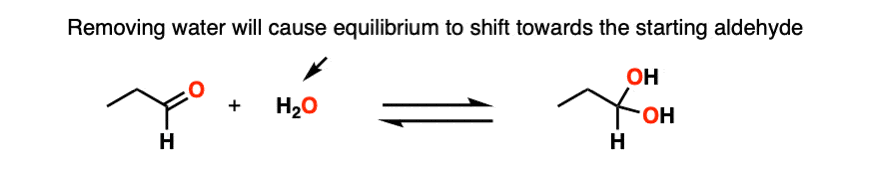
Note 3 – Le Chatelier’s principle, roughly stated, says that changing the concentration of a chemical will shift the equilibrium to the side that would counter that change in concentration. So if we reduce the concentration of CH3OH in the reaction below (i.e. by removing solvent) the reaction would undergo a shift to the left (production of more CH3OH) and thus loss of the hemiacetal.
Note 4 – Attempting to convert carbonyl group of an ester RCO2R into an “acetal” through using ROH / H+ to make RC(OR)3 does not work. However, these species, known as “ortho-esters” have been made through other means (e.g. basic hydrolysis of chloroform).
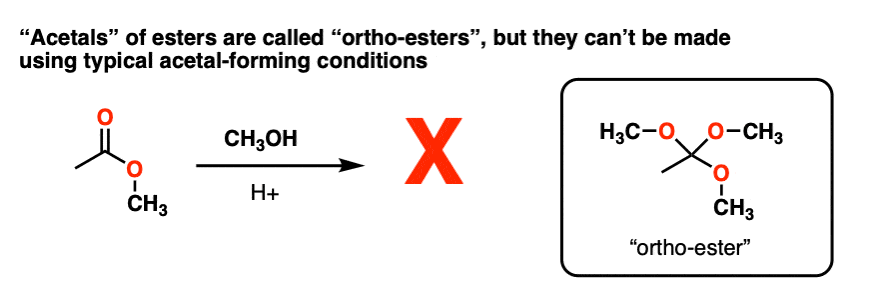
Note 5 – we could also protonate the OR group to give ROH(+), which could be eliminated to give a protonated carbonyl.
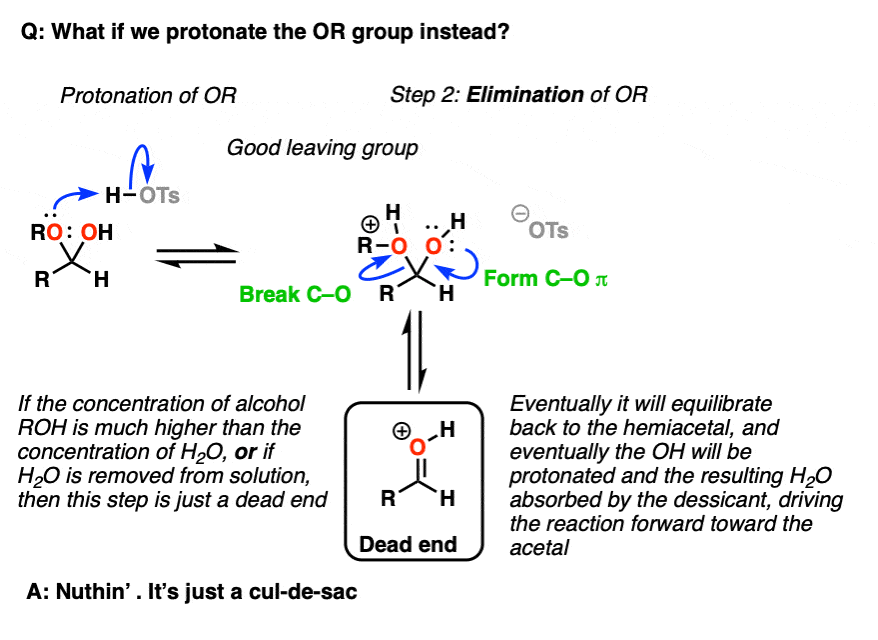
But now ask, “what happens next?”. If the concentration of ROH >> the concentration of H2O [normally the case in these situations] then the protonated carbonyl is just a dead end. Sequestering the H2O as it is eliminated will result in driving the equilibrium towards the final acetal product.
Note 6 – This species is known as an “oxocarbenium ion“.
Note 7 – You could also look at this the opposite way – aldehydes/ketones as a great protecting group for diols!
Note 8 – Here’s a classic example. Treating this molecule with acid results in a bicyclic molecule known as a “spiroketal”.
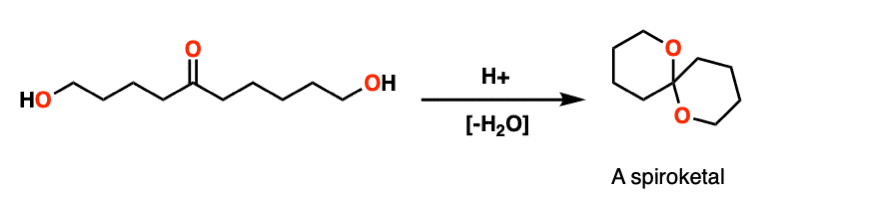
Note 9 – Here’s one way of looking at it. Trade the route from aldehyde to acetal – you should get PADPEAD. Now trace the route from acetal to aldehyde (from right to left). You should get PEADPED.
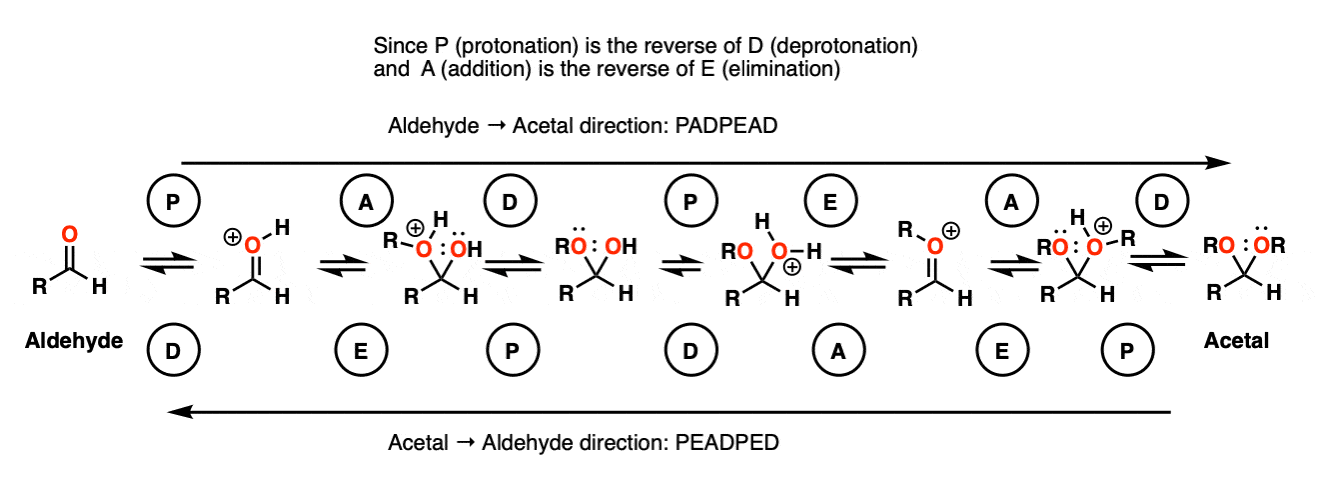
Each mechanism has two P steps and two D steps (no net change in charge – they cancel)
Acetal formation (left to right) has two addition steps and one elimination step.
Aldehyde formation (right to left) has one addition step and two elimination steps.
Quiz Yourself!
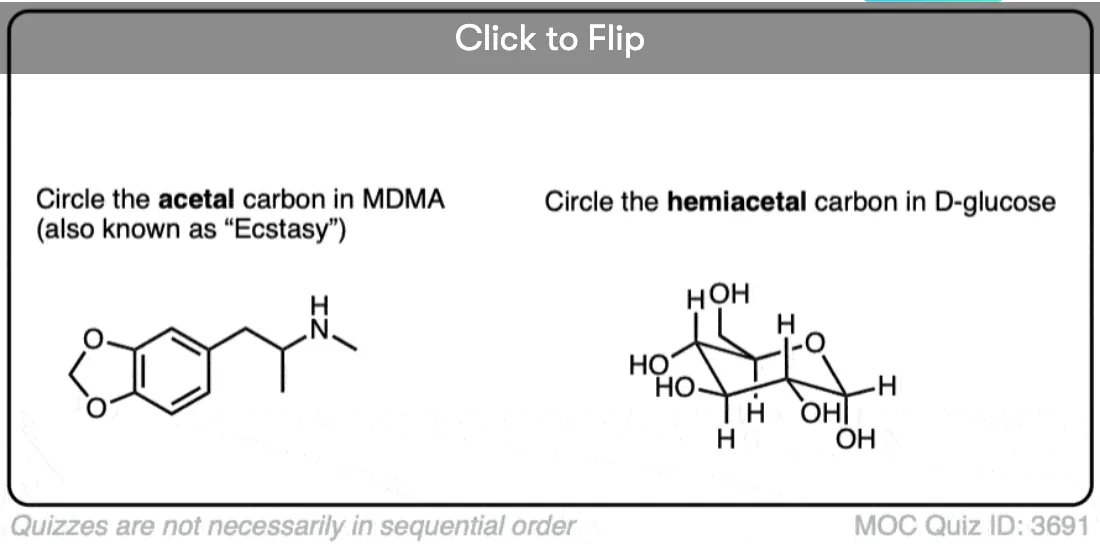
Become a MOC member to see the clickable quiz with answers on the back.

Become a MOC member to see the clickable quiz with answers on the back.

Become a MOC member to see the clickable quiz with answers on the back.
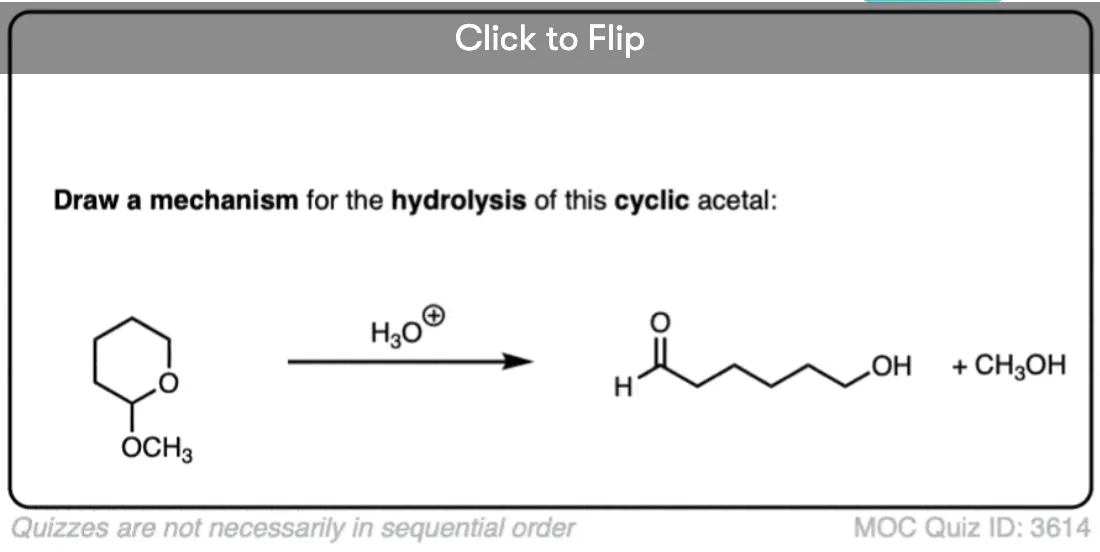
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.

Become a MOC member to see the clickable quiz with answers on the back.

Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading
Acetal formation:
- Darstellung der Acetale
Emil Fischer and Georg Giebe
Ber. 1897, 30 (3), 3053
DOI: 10.1002/cber.189703003121
One of the earliest references in the chemical literature on acetalization reactions, by the legendary chemist Emil Fischer. - —The direct acetalisation of aldehydes
Robert Downs Haworth and Arthur Lapworth
J. Chem. Soc., Trans., 1922, 121, 76-85
DOI: 10.1039/CT9222100076
Haworth and Lapworth are both great chemists in their own right, and in this paper they describe the synthesis of acetals of a variety of common aldehydes (e.g. cinnamaldehyde, anisaldehyde, citral, and others). This chemistry is of significance in the fragrance industry – a lot of aliphatic aldehydes have pleasant odors, but are not stable in either acid or base. The development of methods to protect aldehydes that can maintain their odor and structure is therefore commercially significant. - Methods for the Preparation of Acetals from Alcohols or Oxiranes and Carbonyl Compounds
Frans A. J. Meskens
Synthesis 1981; 1981(7): 501-522
DOI: 1055/s-1981-29507
Review on acetal-forming reactions with a variety of references. - ACETAL
Homer Adkins and B. H. Nissen
Organic Syntheses, Vol. 1, p.1 (1941); Vol. 3, p.1 (1923)
DOI: 10.15227/orgsyn.003.0001
An early procedure in Organic Syntheses for the preparation of acetaldehyde diethyl acetal. This reagent can be used as a convenient, easier-to-handle, less volatile source of in situ acetaldehyde.Acetal Hydrolysis: - Mechanism and catalysis for hydrolysis of acetals, ketals, and ortho esters
G. H. Cordes and H. G. Bull
Chemical Reviews 1974, 74 (5), 581-603
DOI: 10.1021/cr60291a004
A review on the work that had been done up to that point on determining the mechanism of acetal hydrolysis. Several studies point to carbocation formation as the rate-determining step. - Acid-catalyzed Hydrolysis of Acetal and Chloroacetal
Maurice M. Kreevoy and Robert W. Taft Jr.
Journal of the American Chemical Society 1955, 77 (11), 3146-3148
DOI: 1021/ja01616a075
This paper demonstrates the relationship between the rate of hydrolysis and the Hammett acidity (Ho) of the solvent, a general acidity function. This gives further support for the hypothesis that carbocation formation is the rate-determining step. - Trapping of the oxocarbonium ion intermediate in the hydrolysis of acetophenone dimethyl ketals
P. R. Young and W. P. Jencks
Journal of the American Chemical Society 1977, 99 (25), 8238-8248
DOI: 10.1021/ja00467a019
In this paper, the oxocarbonium ion formed during acetal hydrolysis is trapped with bisulfite ion, establishing that it is indeed an intermediate in the reaction. - Effect of Alkyl Group Size on the Mechanism of Acid Hydrolyses of Benzaldehyde Acetals
Alexanders T. N. Belarmino, Sandro Froehner, Dino Zanette, João P. S. Farah, Clifford A. Bunton, and Laurence S. Romsted
The Journal of Organic Chemistry 2003, 68 (3), 706-717
DOI: 1021/jo0202987
This paper suggests that the mechanism of hydrolysis changes from specific-acid catalyzed (by H+) to general-acid catalyzed depending on the acetal structure. - General acid catalysis in the hydrolysis of 1,3-dioxolanes and 1,3-oxathiolanes. The hydrolysis of acetals and thioacetals of p-(dimethylamino)benzaldehyde
Thomas H. Fife and R. Natarajan
Journal of the American Chemical Society 1986, 108 (9), 2425-2430
DOI: 1021/ja00269a048 - General acid catalyzed acetal hydrolysis. The hydrolysis of acetals and ketals of cis- and trans-1,2-cyclohexanediol. Changes in rate-determining step and mechanism as a function of pH
Thomas H. Fife and R. Natarajan
Journal of the American Chemical Society 1986, 108 (25), 8050-8056
DOI: 10.1021/ja00285a028
These two papers demonstrate that as expected, the mechanism of acetal hydrolysis varies with pH. - General acid catalysis of acetal, ketal, and ortho ester hydrolysis
Thomas H. Fife
Accounts of Chemical Research 1972, 5 (8), 264-272
DOI: 1021/ar50056a002
An account by Prof. Fife on his work relating to the mechanisms of acetal hydrolysis. - Intramolecular Catalysis in the hydrolysis of glycosides and acetals
B. Capon, M.C. Smith et. al.
J. Chem. Soc. B. 1969, 1038-1047
DOI: 10.1039/J29690001038
Example of an acetal hydrolysis reaction where the rate is greatly accelerated due to an intramolecular proton transfer. - The Evaluation of Inductive and Resonance Effects on Reactivity. I. Hydrolysis Rates of Acetals of Non-conjugated Aldehydes and Ketones
Maurice M. Kreevoy and Robert W. Taft Jr.
Journal of the American Chemical Society 1955 77 (21), 5590-5595
DOI: 10.1021/ja01626a040
Measures reaction rates for hydrolysis of a variety of acetals and ketals. Table below:

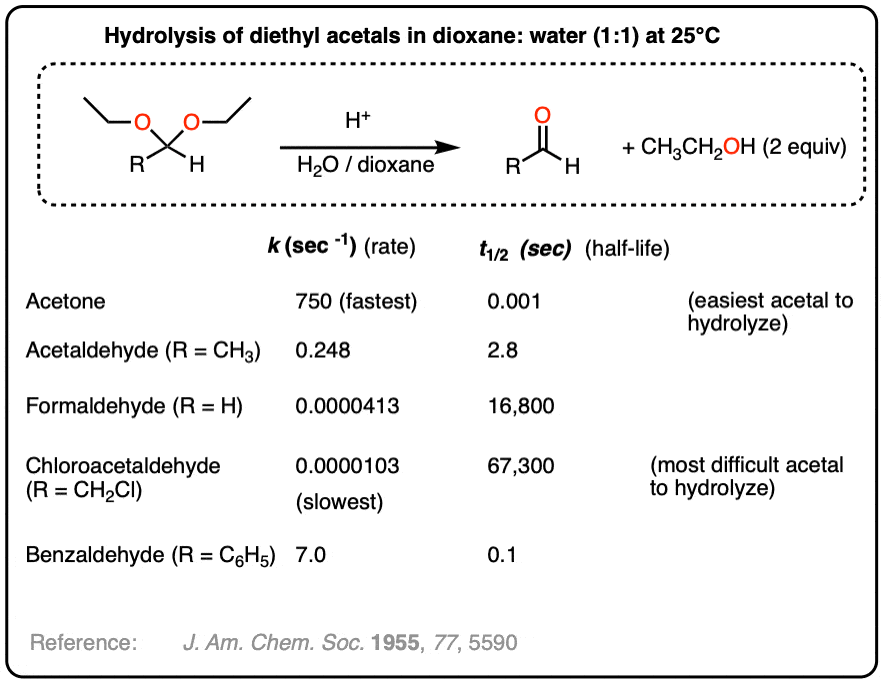
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Hello. I need help to draw the mechanisms of a- glucose from acyclic glucose under acidic conditions and b – glucose from acyclic glucose under basic conditions. Could you help me, please?
Sir,
In the section of formation of acetal from aldehyde and ketones there is a typo error!!!
“”Going from an acetal to a hemiacetal is P A D. “””
Instead it should have been
“” going from an aldehyde to a hemiacetal is PAD “”.
Great catch. Thank you!
I especially liked the PADPEAD mnemonic, it makes remembering the mechanism steps so much easier. The part about cyclic hemiacetals and how equilibrium plays into their stability really helped me understand why some reactions favor ring formation. If anyone wants to dive deeper or get extra help with chemistry concepts like this, there are some excellent tutors available here: [redacted by admin]
Can phenol and formaldehyde give hemiacetal in basic conditions?
Yes, the phenol oxygen can add to formaldehyde to give a hemiacetal-like structure.
I think the spiroketal (Note 8) is formed from the starting ketodiol that should have one CH2- less (totally 9 carbons).
Can hemiacetals and acetals be hydrolysed in alkaline medium?
In question – 0402 can it be this way
First the Oxygen atom attack the H+ in the medium an leave as an alcohol leaving an sp2 hybrid carbocation species.
Then the ethan-1,2-diol attack that carbon resulting the oxygen to take the pi electrons of the double bonds and become negative which later can form an -OH group due to H+ in the medium
The carbon is still positively charged then gets an intramolecular attack from the other carbonyl oxygen atom
This will give us a product will give a product having a 6 atom heterogenous ring(as it will have an oxygen atom) and the ring will have an hemi-acetal group.
I would be really grateful if you tell me where am I wrong an point out my mistakes
Thank you!!
P.S- I assume in my solution there are too many intermediates in the reaction mechanism which can result in making it less favorable or due to hinderance
If there is more to it please let me know
If you have an OH on a carbonyl compound and add alcohol to it in acidic conditions, does it make a difference which OH attacks first? In other words, will the inter or intramolecular reaction proceed first?
Thank you!
Would need to see the specific example, but generally, if a 5- or 6- membered ring can be formed, you can assume that will happen first.
For example, sugars like glucose have an aldehyde at C-1 and alcohols at the other 5 carbons, and they tend to spend most of their time (99%) as the cyclic hemiacetal. See here:
https://www.masterorganicchemistry.com/2017/07/13/pyranoses-and-furanoses-ring-chain-tautomerism-in-sugars/
in reaction of acetone with phenol in presence of conc H2SO4 ,electrophilic addition occurs and acetal do not form why ?
Strong acid is probably protonating the phenol. Don’t think phenol forms acetals.
Can you explain how acetals or hemiacetals get converted to aldehyde in acidic medium.
I didnt quite understand how protonation makes the carbonyl more electrophilic in acidic condition. Could you please explain more about that part?
google should definitely rank this page higher than “chem.libre textbooks” for someone searching “acetal vs hemiacetal formation.”
I automatically move past chem.libre and come here…. this page is The Go To Source for All Things O Chem. Thank you guys.
Appreciate it Eric. How’s tutoring?
Hello sir, thank you so much for these great notes.
My doubt is – In the 3rd bit of the 3rd question how can we decide which OH should be used first, why didn’t you use the other OH first?
Thank you.
Thanks, this is well simplified and amazing
what is more poler keton or ketal
Ketone will have a larger dipole moment.
It can form an acetal only with the carbohydrate, without an addition of an alcohol group.
Thank you very much in advance!
Helped a lot!
Tem como formar um acetal apenas com o carboidrato, sem a adição de um grupo álcool.
Desde já, agradeço!
Ajudou muito!
“… treatment of glucose with a reducing agent like NaBH4 will eventually completely reduce it to the alcohol, hence its classification as a reducing sugar.”
I don’t think that’s right. It is the fact that it can be oxidized to the carboxylic acid, and reduce another compound in the process, that makes it a reducing sugar.
You are correct. My mistake. https://www.masterorganicchemistry.com/2017/09/12/reducing-sugars/
Could you include formation of hemiacetal under basic conditions? Isn’t that the only non-neutral pathway that would work?
Is it correct that this process of making an acetal won’t work on an ester? If not, why not?
I have a lactone with a ketone, and I’m trying to hydrolyze it with LiAlH4, then PBr3 and Mg to close it up as an all carbon ring, but with the ketone. It seems though that if I add either an acetal or acid the ester should open?
No, it won’t work on an ester. You’ll just end up doing transesterification. There are such things as ortho-esters, but they are not formed from esters. https://en.wikipedia.org/wiki/Orthoester
Just wondering, does the pH of the solution affect the equilibrium between the hemiacetal and aldehyde form of glucose?
Yes – it makes the OH into O- which is a better nucleophile (and worse leaving group) than OH, and it will increase the amount of ring-closed product.
Great explanation. The University lost a great professor
Quick question, in your structure of glucose, I’ve never seen the OH groups on Carbons 3 & 4 drawn that way. It actually looks like you’ve drawn the sugar Gulose there. Am I missing something? Cheers.
Fixed. Thanks!
Wondering if you could post something explaining just how to look at one and tell if it is a hemiacetal or acetal. I understand that the hemi has an alcohol and an ether attached to the same carbon. But it would be nice to see each part of it labeled out and explained to us. Thanks this infor was amazing!
I put a box around the relevant atoms that make up the acetal and hemiacetal functional groups. Does this help at all, or does it make it look worse?