Alcohols, Epoxides and Ethers
Thiols And Thioethers
Last updated: May 28th, 2026 |
Thiols and Thioethers: Properties and Key Reactions
If you can get beyond their foul smells, thiols have a lot of similar characteristics to alcohols!
- Like alcohols, they can be deprotonated with base and undergo SN2 reactions with alkyl halides to give thioethers (sulfides).
- Thiols tend to be considerably more acidic than alcohols since the sulfur atom is more polarizable.
- Their conjugate bases, “thiolates” are excellent nucleophiles.
- Thiols can undergo oxidation to give disulfides.
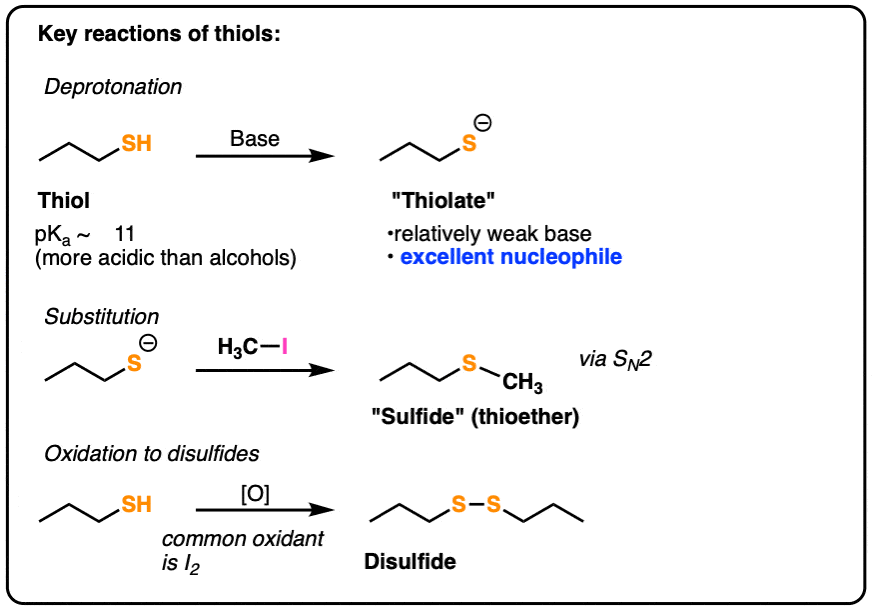
Table of Contents
- Thiols and Thioethers
- First Things First: Thiols Smell Bad
- Thiols Are More Acidic Than Alcohols
- Sulfur: Less Basic, More Nucleophilic Than Oxygen
- Common Reactions of RS(–) : SN2 Reactions With Alkyl Halides
- Key Differences Between Thiols and Alcohols
- Summary: Thiols and Thioethers
- Notes
- Quiz Yourself!
- (Advanced) References and Further Reading
1. Thiols and Thioethers
One of the most powerful insights from learning the periodic table is the fact elements in the same period (column) have similar chemical properties.
For instance
- Alkali metals (Li, Na, K…) all shed an electron relatively easily to form monovalent cations
- Halogens (F, Cl, Br) all gain an electron relatively easily to form halide ions.
- Noble gases (He, Ne, Ar, Kr…) tend to be chemically inert.
So how does what we’ve learned about the reactivity of oxygen (O) based functional groups translate to its heavier cousins sulfur (S) , selenium (Se) and tellurium (Te)?
Specifically, how does the chemistry of alcohols (ROH) and ethers (ROR) compare with the chemistry of thiols (RSH) and thioethers [aka sulfides] (RSR)?.
Great question! That’s the topic of today’s post, specifically thiols and thioethers. We won’t really talk about selenium chemistry beyond briefly mentioning its acidity, and we definitely won’t go anywhere near tellurium chemistry, following Wöhler’s advice on diethyl tellurium that “its obnoxious and persistent smell is connected to unpleasantness, which one wouldn’t like to endure a second time“, and that “the smell is so persistent, that one has to avoid social life for several months in order not to molest other people“.
Let’s start with what’s similar between alcohols/thiols, ethers/thioethers, and then move to what’s vastly different.
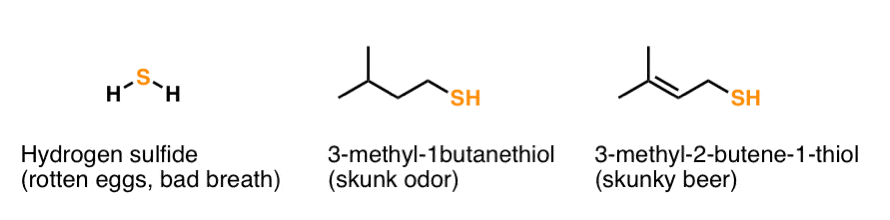
2. First Things First: Thiols Smell Bad
Open a textbook that discusses thiols and one of the first things they’ll mention is their vile stench. We wouldn’t want to break with this honourable tradition here at MOC.
Hydrogen sulfide (H2S) is responsible for the smell of rotten eggs and bad breath, among other unpleasant things. Thiols contribute to the smell of skunks and “skunky” beer.
During my time in grad school one of the world’s leading organosulfur chemists had a lab across the hall. These were not people you wanted to be downwind from. Imagine getting on the city bus and having the people next to you get up from their seats and move down to the other end. Those stories were not uncommon from the people in that lab.
BTW: the sulfur chemists’ best friend is bleach (NaOCl), which oxidizes stinky thiols to relatively odourless sulfoxides (see #5, below).
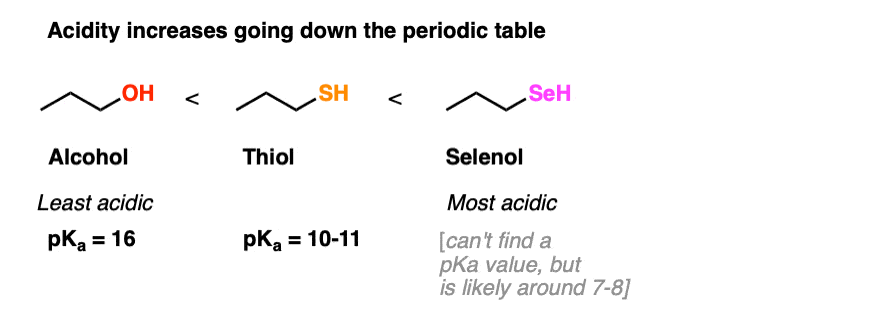
3. Thiols Are More Acidic Than Alcohols
We’ve seen that alcohols are relatively acidic (pKa‘s of about 16-17). Thiols are more acidic than alcohols by an average of about 5 pKa units or so ( pKa of about 11 for the thiol pictured below). Remember that pKa is logarithmic, so that means they’re about 105 times more acidic.
Why might that be?
When understanding acidity trends it greatly helps to think about the stability of the conjugate base
Recall that acidity will be increased by any factor which stabilizes the conjugate base. (See post: 5 Key Factors That Influence Acidity)
[Since acid-base reactions are equilibria, if we stabilize the product (conjugate base) we therefore make the equilibrium where the starting material (acid) loses a proton more favourable, increasing Ka. In other words, we increase the acidity.]
In the case of sulfur, the negative charge will be borne on a larger atom (sulfur > oxygen) and that negative charge dispersed over a greater volume. Greater volume = more diffuse charge = greater stability. (See post: 7 Factors That Stabilize Negative Charge in Organic Chemistry)
The greater acidity of the thiol is indicated by the lower measured pKa of the thiol pictured above (about 10-11) relative to alcohols (pKa 16-18)
The same holds true for selenium, which has an even larger atomic radius. I can’t find a pKa value for propaneselenol but would estimate it to be about 7-8. [If someone has a better number, please let me know!]
A corollary of the fact that the conjugate base is more stable is that thiolates [the conjugate bases of thiols] are weaker bases than alkoxides. [“The stronger the acid, the weaker the conjugate base”]. (See post: How to Use a pKa Table)
4. Sulfur: Less Basic, More Nucleophilic
Remember how alkoxides [RO–] can act as nucleophiles in SN2 reactions as well as the base in elimination (E2) reactions? (See post: Williamson Ether Synthesis – Planning)
With thiolates [RS–], E2 reactions aren’t an issue. The weaker basicity of thiolates means that only SN2 reactions occur with alkyl halides.
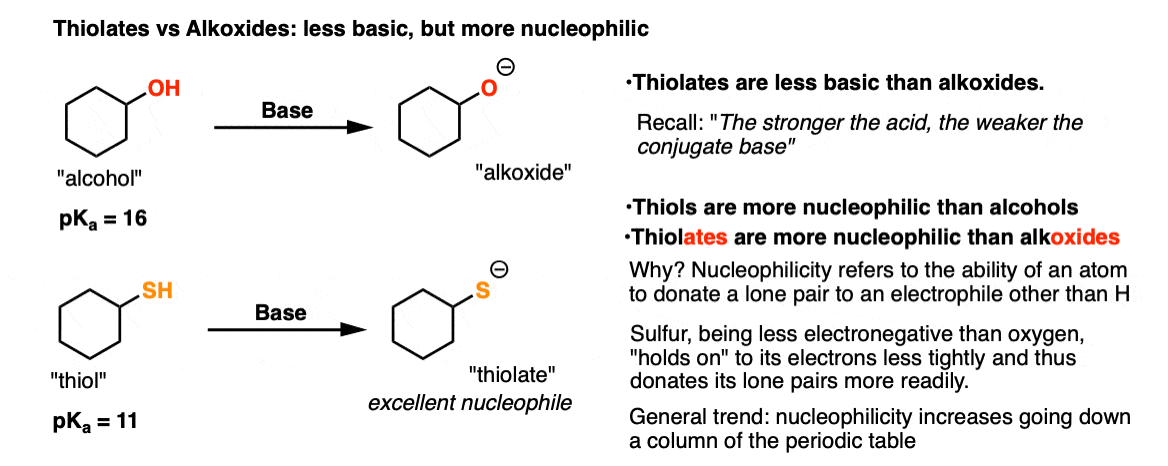
Thiols are more nucleophilic than alcohols, and thiolates are more nucleophilic than alkoxides. Since nucleophilicity is measured by reaction rate, that means that these sulfur nucleophiles tend to react faster with typical electrophiles (like alkyl halides) than their oxygen-based cousins.
Why? As we’ve discussed before, the lower electronegativity of sulfur (relative to oxygen) means that its pairs of electrons are held onto less tightly than oxygen, making them more readily available for donation to electrophiles (like alkyl halides).
This trend continues as we descend a column of the periodic table, so in general, nucleophilicity increases in this direction as well (i.e. RSe– is even more nucleophilic than RS– ).
5. Common Reactions of Thiolates: SN2 Reactions
We’ve seen that one of the most important reactions to form ethers is the Williamson ether synthesis. (See post: The Williamson Ether Synthesis)
Here’s a textbook example of the sulfur analogue of this reaction: substitution of alkyl halides with thiolates to form thioethers (also known as sulfides).
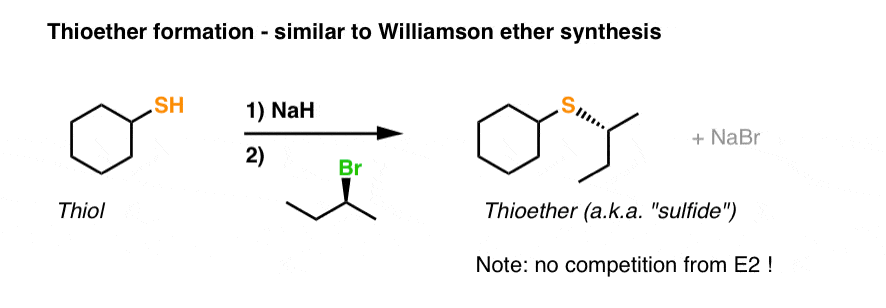
This process resembles the Williamson to a tee.
- First, a strong base deprotonates the thiol (we use NaH here, but many other bases could also be used).
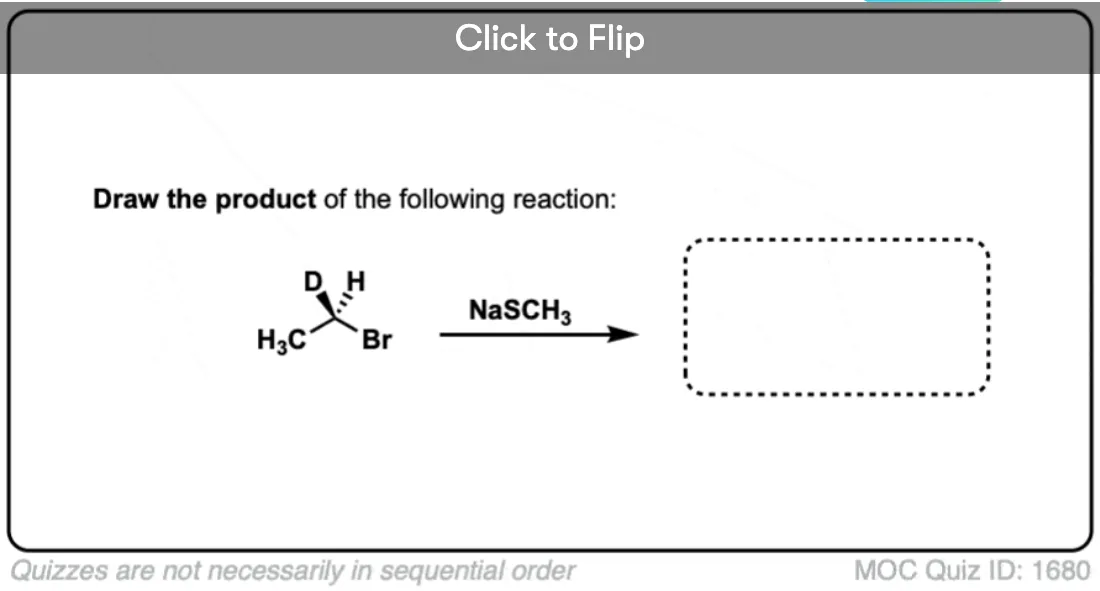
- Secondly, we add an alkyl halide, and an SN2 reaction results in formation of S-C and breakage of C-Br with inversion of stereochemistry.
As mentioned above, note that if we were using an alkoxide, we’d have to worry about the E2 reaction competing with the SN2. With thiolates, it’s not an issue due to their decreased basicity.
Since it comes up so much in exams, the intramolecular version is important to note. You’ll see that we’re still forming C–S here and breaking a carbon-halide bond. Don’t forget that inversion occurs on the stereocenter on the secondary carbon!
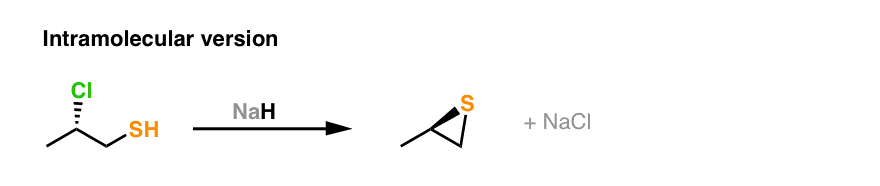
6. Key Differences Between Alcohols And Thiols
A. Oxidation
In previous posts we saw that primary alcohols are oxidized to aldehydes and secondary alcohols are oxidized to ketones (See post: Oxidation of Alcohols)
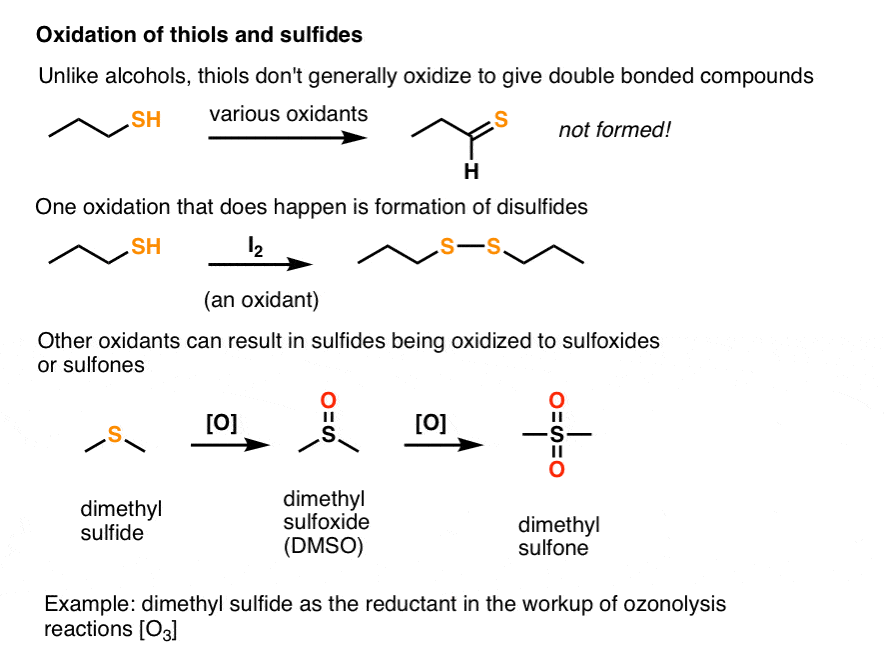
This doesn’t work with thiols! Thiols aren’t oxidized to π bonds in the same way. The C–S π bond is actually quite weak due to poor orbital overlap. [Note 1]
Instead, two different types of oxidation reactions occur with thiols.
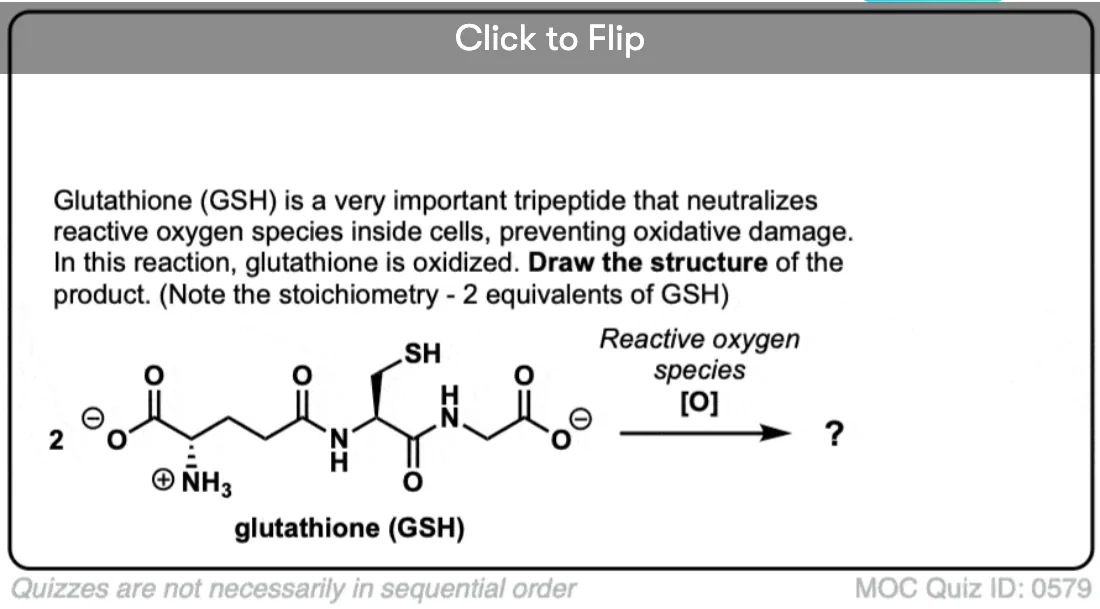
- Thiols can be oxidized to disulfides through treatment with a mild oxidant like iodine (I2).
- A second oxidation pathway involves oxidation of sulfides to sulfoxides and sulfones through treatment with oxidants such as O3 (ozone) and peroxyacids such as m-chloroperoxybenzoic acid (m-CPBA). Note that sulfur can exceed an octet of electrons whereas oxygen cannot.
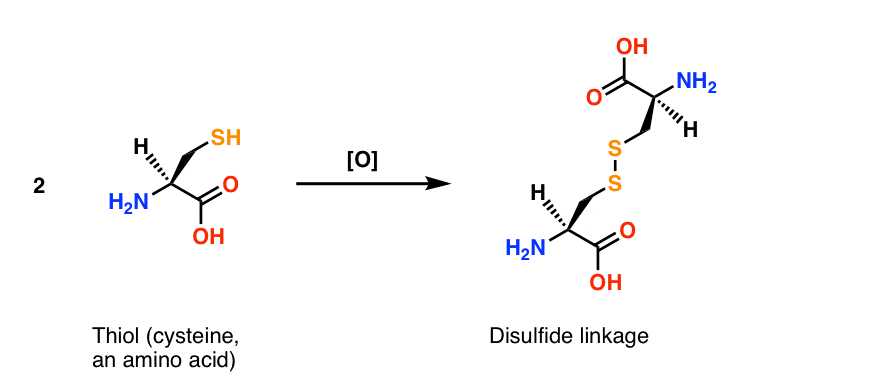
The oxidation of thiols to disulfides has important biological implications. The important amino acid cysteine contains a thiol group and disulfide bonds are responsible for the tertiary structures of proteins. They act like “hooks” that help to hold disparate parts of the peptide chain together.
B. Hydrogen Bonding Isn’t As Significant With Thiols
The electronegativity of sulfur is only 2.6 whereas hydrogen is 2.2. That means there isn’t much of a dipole in the S-H bond, and hydrogen bonding is not significant. Contrast H2O, with a boiling point of 100°C, with H2S and its boiling point of –60°C. Intermolecular forces are not very strong!
C. Reactions With Acids Are Slightly Different
In this series on alcohols and ethers we have seen countless examples of treating an alcohol or ether with H+ and converting it to its conjugate acid, making a better leaving group in the process.
This isn’t as significant for thiols. The S-H bond is significantly weaker than the O-H bond [81 kcal/mol vs. 109 kcal/mol].
Secondly, due to the smaller dipole (electronegativity difference) of the S-H as opposed to O-H, there is less partial negative charge on sulfur and therefore less electrostatic attraction between S and the H of various acids.
A better way of converting sulfur to a good leaving group is by treating it with Lewis acids such as Hg(OAc)2 . We haven’t really covered Hard-Soft Acid Base (HSAB) theory here on MOC (which is an advanced topic) but in order to further understand the differences in reactivity of O and S with various acids, the linked Wikipedia article is a useful primer.
D. There are no “thio” versions of many reactions of alcohols
We’ve seen many interesting reactions that have led to alcohols and epoxides. For example, treatment of alkenes with OsO4 results in vicinal diols. It’s tempting to think that there might be an analogous reaction between alkenes and OsS4 to give vicinal dithiols. There isn’t (and it’s not for lack of trying). Nor is there a sulfur equivalent to the formation of epoxides from alkenes with, say, the sulfur equivalent of mCPBA. So while moving down a column of the periodic table can sometimes provide useful inspiration for new reactions, often – for unclear reasons – it does not.
7. Summary: Thiols and Thioethers
In the next post (our last on alcohols) we’ll tie all the reactions we’ve learned together into a “reaction map”.
Next Post: Synthesis (6) Reactions of Alcohols
Notes
Note 1. In grad school a visiting speaker gave a talk on making thioaldehydes and thioketones. I missed it, but an emeritus professor who had a hood in our lab was able to attend. He reported back: “What I learned from today’s seminar about the C=S bond…. is that you don’t want to make compounds with C=S bonds”, he said with a laugh. [They’re quite unstable]
[Caveat: This applies to thioaldehydes/thioketones. Molecules like thioamides or thioureas where an atom like N or O are adjacent to the carbon bearing C=S are generally reasonably stable]
Note 2: Relevant video for this post.
Quiz Yourself!
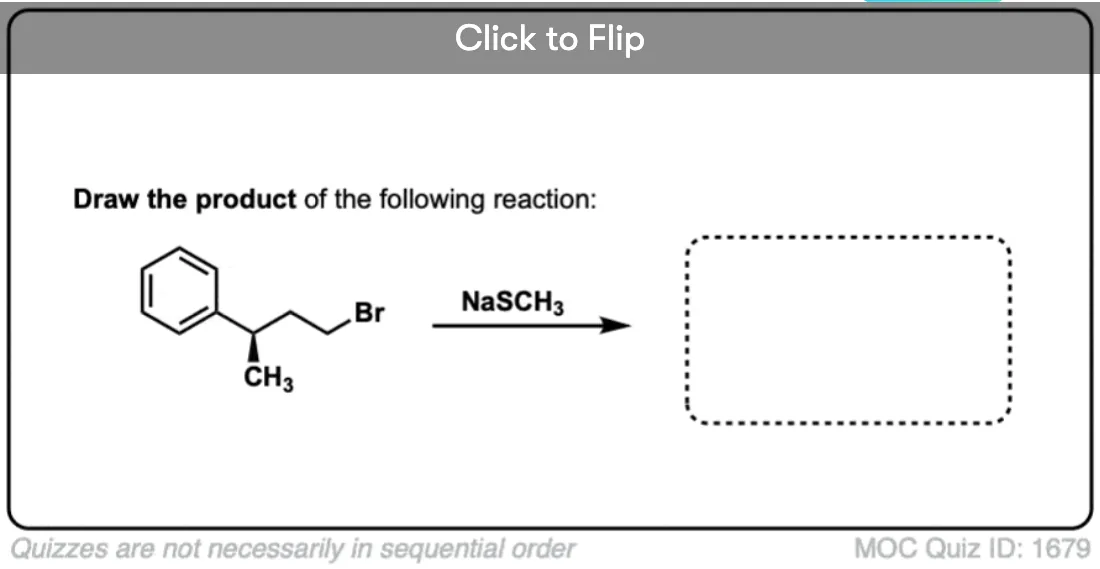
Become a MOC member to see the clickable quiz with answers on the back.
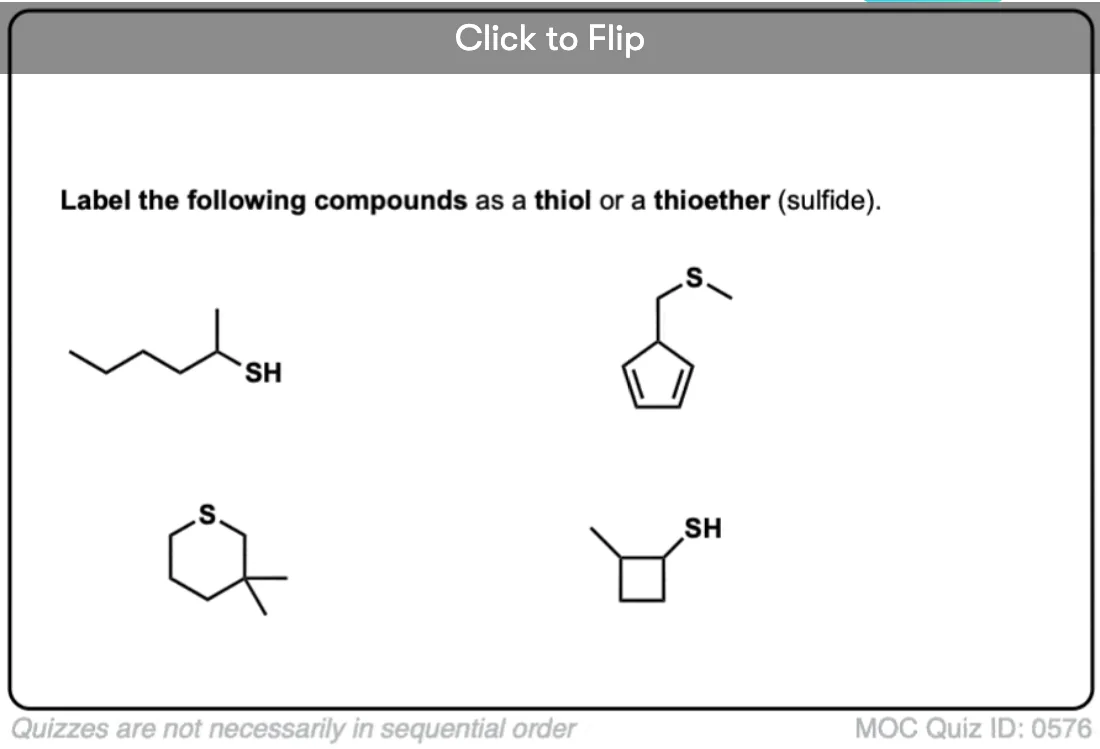
Become a MOC member to see the clickable quiz with answers on the back.
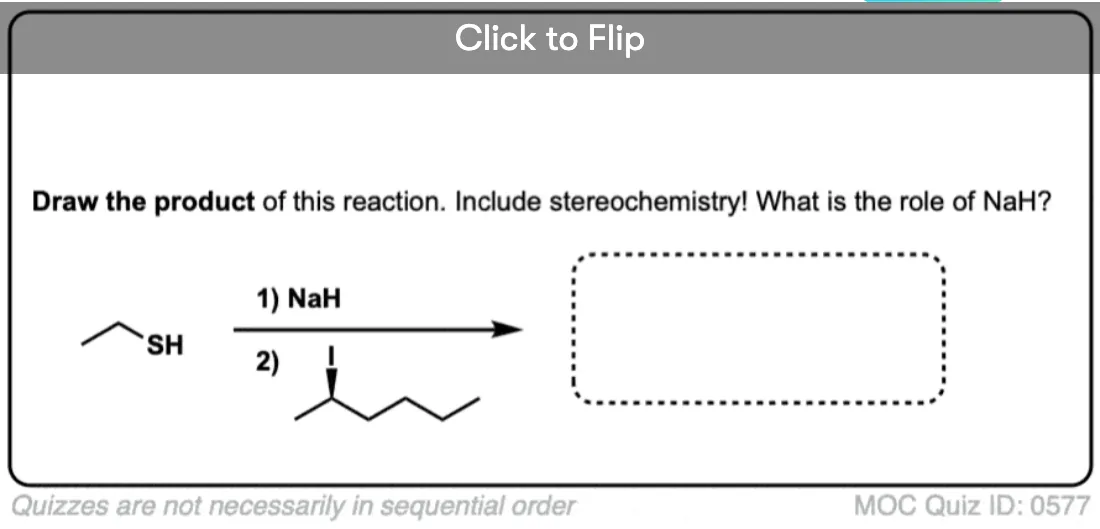
Become a MOC member to see the clickable quiz with answers on the back.
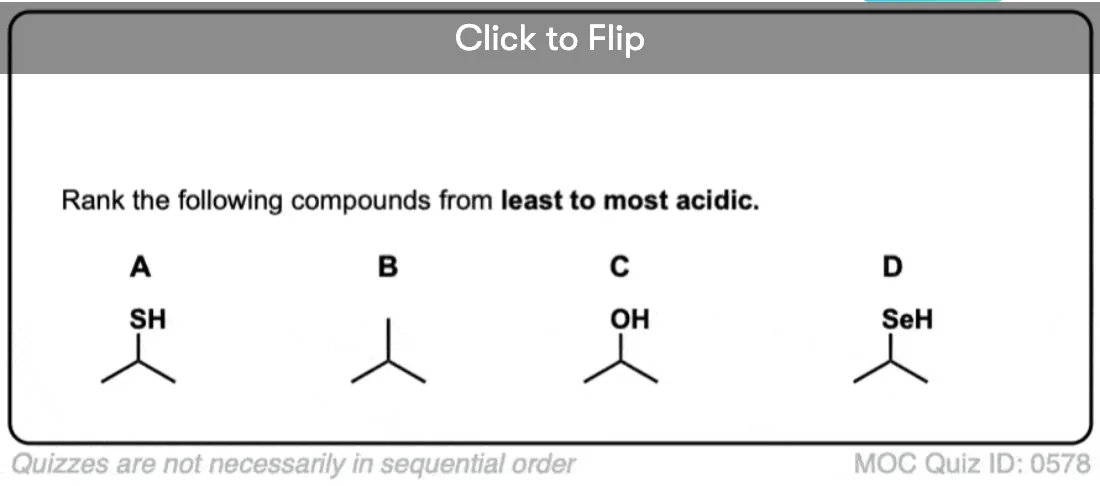
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.

Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading
- Acidic dissociation constants of thiols
James P. Danehy and K. N. Parameswaran
Journal of Chemical & Engineering Data 1968, 13 (3), 386-389
DOI: 1021/je60038a025
This contains a list of pKa’s for over 100 thiols. Very useful reference. - Synthesis of thiols, sulfides, sulfoxides and sulfones
Christopher M. Rayner
Synth. 1995, 2, 409-440
DOI: 10.1039/CO9950200409
Nice review on sulfur chemistry in organic synthesis.Notable medicinal chemist Derek Lowe has a series of posts titled “Things I won’t work with” on his (excellent) blog. Two of the compounds below make the list because of their vile smells – thioacetone (CH3CSCH3) and selenophenol (PhSeH). - “Things I Won’t Work With: Thioacetone”. [Link]
- “Things I Won’t Work With: Selenophenol”. [Link]
Amusing articles on the astonishingly powerful odor of thioacetone and (to a lesser extent) selenophenol, from Derek Lowe’s “In The Pipeline” blog.Sulfur atoms on an adjacent carbon can ‘assist’ with reactivity. This is known as ‘Neighboring Group Participation’ or ‘Anchimeric Assistance’. This was first observed by the famed chemist Prof. Saul Winstein (UCLA), who made fundamental contributions to Physical Organic Chemistry. - Kinetics of Hydrolysis and Displacement Reactions of β,β′-Dichlorodiethyl Sulfide (Mustard Gas) and of β-Chloro-β″-hydroxydiethyl Sulfide (Mustard Chlorohydrin)
Paul D. Bartlett and C. Gardner. Swain
Journal of the American Chemical Society 1949, 71 (4), 1406-1415
DOI: 10.1021/ja01172a076 - The Hydrolysis of Bis(2-chloroethyl) Sulfide (Sulfur Mustard) in Aqueous Mixtures of Ethanol, Acetone and Dimethyl Sulfoxide
Ian Tilley
Aust. J. Chem. 1993, 46, 293-300
DOI: 10.1071/CH9930293
One of the prime examples of neighboring group participation is the hydrolysis of ‘sulfur mustard’. The sulfur atom allows this molecule to hydrolyze extremely easily (yielding HCl), since it can form a 3-membered episulfonium intermediate. - Hydrolysis of mustard derivatives in aqueous acetone-water and ethanol-water mixtures
Yu Chu Yang, J. Richard Ward, and Thomas Luteran
The Journal of Organic Chemistry 1986, 51 (14), 2756-2759
DOI: 10.1021/jo00364a025 - Neighboring group participation by sulfur involving four-membered-ring intermediates (RS-4)
Ernest L. Eliel and David E. Knox
Journal of the American Chemical Society 1985, 107 (10), 2946-2952
DOI: 10.1021/ja00296a019
Sulfur atoms further out can also act as neighboring groups, geometry permitting.Disulfide bond formation: - Kinetics and mechanism of the oxidation of n-dodecanethiol and pyridine-substituted ethanethiols by iodine molecule in acetonitrile
David L. De Leeuw, W. Kenneth Musker, and Joyce Takahashi Doi
The Journal of Organic Chemistry 1982, 47 (25), 4860-4864
DOI:1021/jo00146a008
This goes over the mechanism of the oxidation of thiols with I2, and includes a mechanism and kinetic rate equation. - Oxidation of cysteine to disulfides.
Sigma-Aldrich Technical Bulletin.
The oxidation of thiols to disulfides is hugely important in biology, due to the naturally occurring amino acid Cysteine. The importance of Cysteine in biochemistry is due to the fact that the -SH group can form disulfide bonds, which is one of the forces leading to the tertiary structure of peptides and proteins. Synthetic peptides with 2 or more Cys residues can be oxidized to give a disulfide bond, and when 3 or more are present, this is something that needs to be done selectively, as the link explains.Due to their even worse smells, organoselenium and -tellurium chemistry has progressed much slower than related thiol chemistry. - Theoretical Calculation of pKas of Selenols in Aqueous Solution Using an Implicit Solvation Model and Explicit Water Molecules
Bishnu Thapa and H. Bernhard Schlegel
The Journal of Physical Chemistry A 2016, 120 (44), 8916-8922
DOI: 10.1021/acs.jpca.6b09520
Theoretical calculations are not the same as experimental determination, but this gives a place to start. - Phenylseleno neighboring group. Solvolysis of 2-phenylselenoethyl chloride
Samuel P. McManus and David Hinkle Lam
The Journal of Organic Chemistry 1978, 43 (4), 650-651
DOI: 10.1021/jo00398a028
Selenium can also engage in neighboring group participation. - Organotellurium Compounds.
Sigma-Aldrich Technical Bulletin.
This publication by Sigma-Aldrich contains a review on organotellurium chemistry.
thank you that was so nice. I would like to know can Thioctic acid and prop-2-ene-1-thiol react with each other and form a new derivative of thionic acid which has a -C3H5SH connecting to carbonyl? i mean from alkyl part
Nice work. One small remark: the functional group R-S-R is called sulfide (with its own name). The name thioethers (as derivatives of ethers) was abandoned more than 25 years ago. If you want to have a neat website, stop using the old term “thioether”. Maybe you can mention “in the old nomenclature…”.
How the thiol radical forms under the extreme conditions like extreme oxidation?
I can’t really answer this question because it is not very precise.
– Which thiol radical?
– What extreme conditions?
– What is meant by extreme oxidation?
– On what substrates?
Do you have an example?
Can two thiolate ions combine together and then further react with Silver ?
Yes, possibly, but if there are any traces of oxygen present, disulfides will form.
is there a reaction/s that can be carried out with hydroxyl groups but not with thiol groups, especially if the two groups are binded to the same molecule?
Thanks,
My suggestion would be to just bite the bullet and selectively protect either the hydroxy or thiol groups. Protection and deprotection are two steps that almost always yield near quantitative yields. It lets you focus on the chemistry you want to do without worrying about functional group compatibility.
You should also mention oxidation R-SH to acid by HNO3, KMno4. And also i am missing here oxidation of disulfides to sulfoxid and sulfon.
Thanks for mentioning that. Yes, not covered here.
May I use your image for key reactions of thiols in my lectures?
Yes, of course.
You remarked that it’s H-bonding isn’t as much as ethers so can we say it is less soluble in water wet ethers or is it completely immiscible???
Thanks in advance!
This looks like a highly informative site. Great work!
Here is the question I dont know whether you have a section for metal complexes but: Would you expect thiols or thioethers to form complex with the copper in chlorophylin?
Cheers
Murat
Not 100% sure, but, copper does form strong complexes with thiols and thioethers.
Excellent post sir……
But my doubt is will strong bases like grignard abstract the hydrogen from the thiol group????
Yes, it absolutely will.
Thanks for the tutorial!
According to Wikipedia, methylselenol has pKa=5.2, compare to methylthiol pKa=8.3 in the same article. Conventionally methylthiol is reported pKa~10.2, so I suspect the first two values may be measured in a solvent other than water. I hope this helps at least a little bit to quantify the relative acidity of organoselenols.
pKa values of 5.2 and 8.3 are definitely measured in water. If you check a biochem book, you are going to also find the same values for side chains of cysteine and selenocysteine. But I do not know what they mean with the “conventional” pKa.
Interesting. I never thought about the lack of analogs. A -CSSH group would be pretty neat.
Thanks for the amazing post! The comparison between the two is really clear and detailed, and the difference between thiols and alcohols too.
Check the text for “vicuna (sic) dithiols”.
Good post, by the way.