Alkene Reactions
Cyclopropanation of Alkenes
Last updated: May 28th, 2026 |
Cyclopropanation of Alkenes
Alkenes can undergo addition reactions from carbenes to give cyclopropanes. There are three main pathways for this reaction that are generally covered in introductory organic chemistry.
- The simplest method is treatment of diazomethane (CH2N2) with light (“photolysis”) to give methylene carbene, which adds to alkenes to form cyclopropanes. This reaction has the downside of being very difficult to control and also uses toxic and explosive diazomethane.
- A robust method which still sees considerable use today is the use of diiodomethane (CH2I2) with zinc-copper couple (a 90:10 alloy of zinc and copper) which forms cyclopropanes with alkenes via a metal “carbenoid” species.
- Haloforms (e.g. CHCl3, CHBr3) when treated with strong base (e.g. KOH or KOt-Bu) will undergo deprotonation followed by loss of a leaving group to give dihalocarbenes. These can also undergo addition to alkenes.
- Cyclopropanation proceeds through a concerted transition state and is stereospecific.
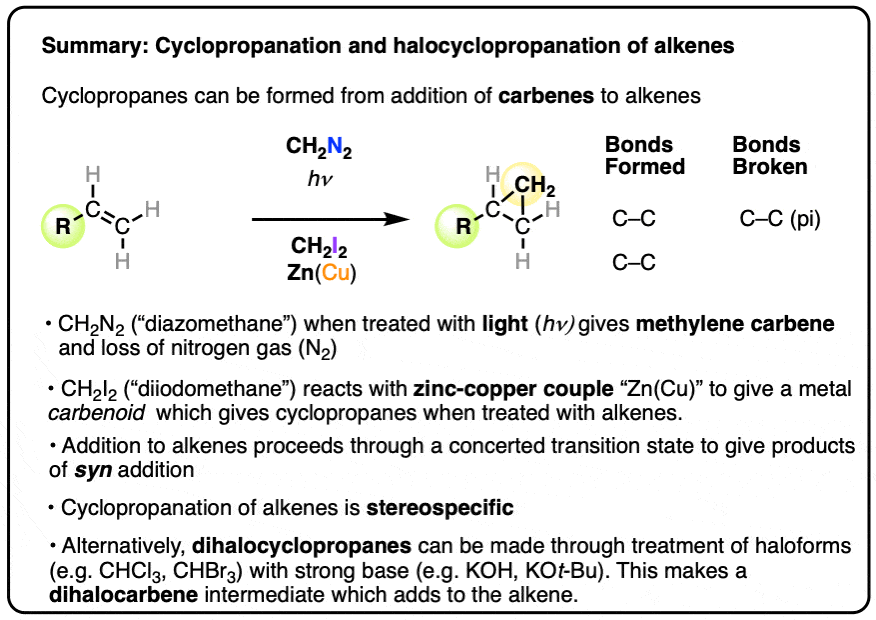
Table of Contents
-
- Cyclopropanation of Alkenes
- Cyclopropanation is Stereospecific
- The Cyclopropanation Mechanism – Photolysis of CH2N2
- The Simmons-Smith Mechanism
- Dihalocyclopropanation of Alkenes
- The Dihalocyclopropanation Mechanism
- Reactions of Cyclopropanes
- Summary
- Notes
- Quiz Yourself!
- (Advanced) References and Further Reading
1. Cyclopropanation of Alkenes
Cyclopropanes are the smallest cycloalkane rings. Due to the fact that their internal angles (60°) are considerably smaller than the ideal tetrahedral bond angle (109.5°), they have considerable (27 kcal/mol) angle strain. (see article: Ring Strain in Cyclopropane and Cyclobutane).
If you’ve ever tried to make a cyclopropane with a molecular model kit, you will quickly discover that the little pieces of plastic will not forgive you for trying to bend them into a constrained triangular shape.
Given this, it might come as a surprise that these unhappy little cycloalkanes can be readily made via addition reactions to alkenes. But they can!
For example, one of the oldest methods for formation of cyclopropanes involves irradiating diazomethane (CH2N2) with visible light (hv) in the presence of an alkene.
In this reaction, two new C-C bonds are formed, a C-C pi bond is broken, and a C-N bond breaks, liberating nitrogen gas (N2).
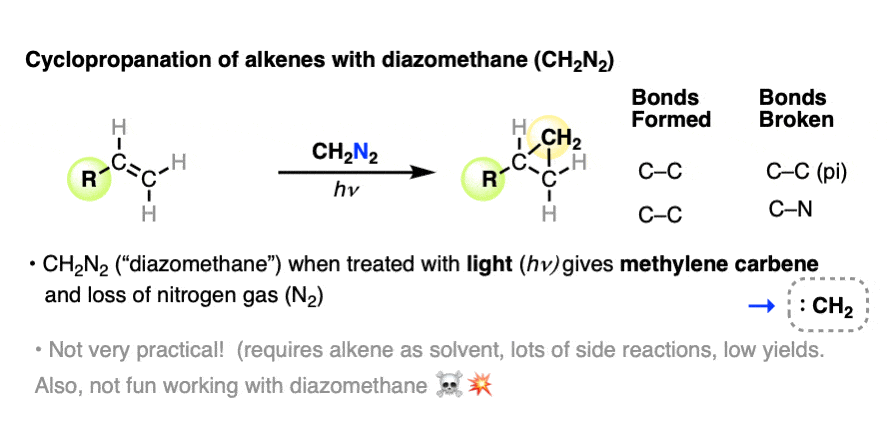
A lot of other side reactions happen too, which we need not get into at the moment. [Note 1 ] Let’s just say that for best results, this reaction is best run using the alkene as the solvent to minimize the many other side reactions that can happen here.
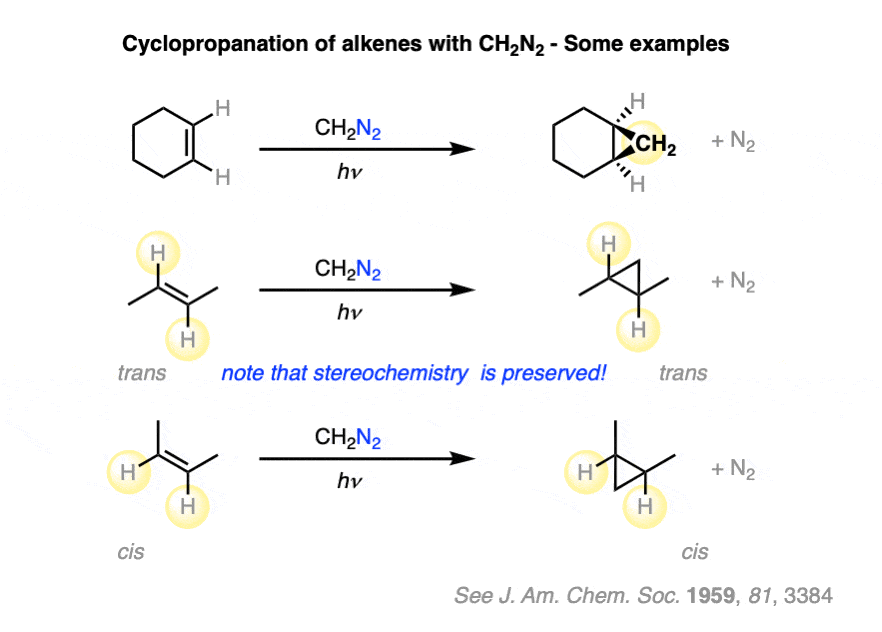
A key observation that has been made about this process is that cyclopropanation occurs such that the stereochemistry about the alkene is conserved. [See Ref]
Diazomethane is the source of many laboratory horror stories and requires special safety precautions for use in the lab. (See article – Diazomethane). It is highly explosive. Dintrogen (N2), an excellent leaving group, is only held on to carbon by a thread, and careless jostling, heat, strong light, or even the rough surface of ground-glass joints can be enough to cause detonation.
And, oh, by the way, it is also highly toxic.
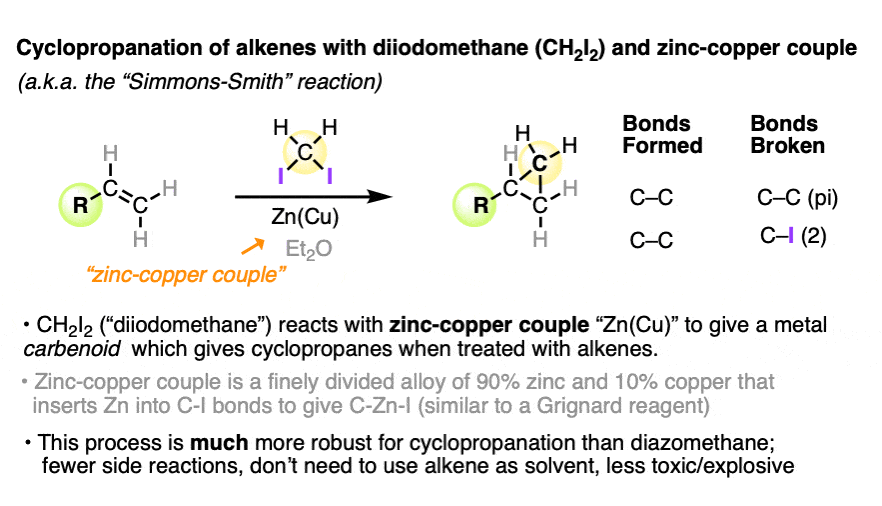
Therefore, it must have been something of a relief in 1958 when two researchers from DuPont in Wilmington, Delaware published a robust method for cyclopropane formation that avoided diazomethane altogether.
Simmons and Smith found that treating diiodomethane (CH2I2) with zinc-copper couple (a 90:10 alloy of finely divided zinc and copper) results in a highly reactive intermediate that smoothly forms cyclopropanes from alkenes.
This reaction, which has come to be known as the Simmons-Smith cyclopropanation, works well with a broad variety of alkenes.
2. Cyclopropanation is Stereospecific
Cyclopropanation of alkenes always results in syn addition of the CH2 to the alkene.
If you try making the model of the anti addition product you will see why. It’s similar to why double bonds can only be cis in small rings; a trans arrangement results in too much ring strain.
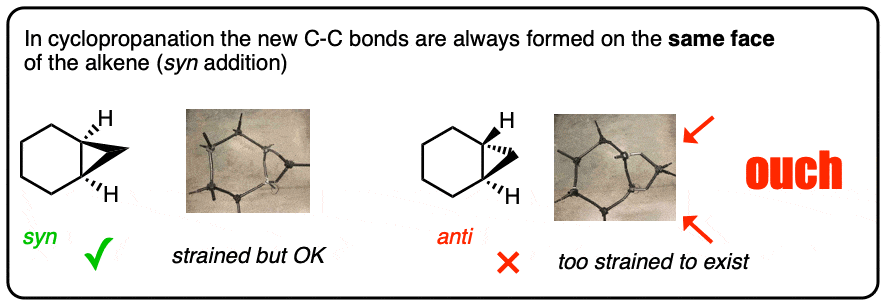
Furthermore, cyclopropanation is an example of a stereospecific reaction. The stereochemistry about the double bond is always conserved. Two groups that are cis to each other on the alkene will be cis to each other on the cyclopropane ring.
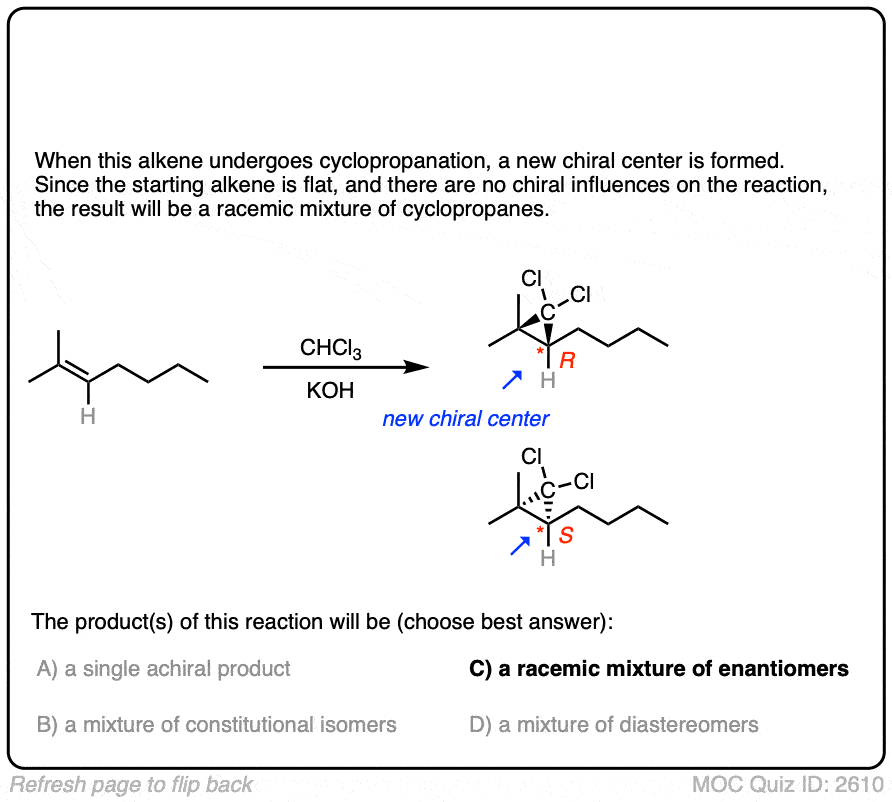
For example, the two alkenes, cis– and trans– hex-3-ene, are stereoisomers that differ only in the configuration at a single carbon.
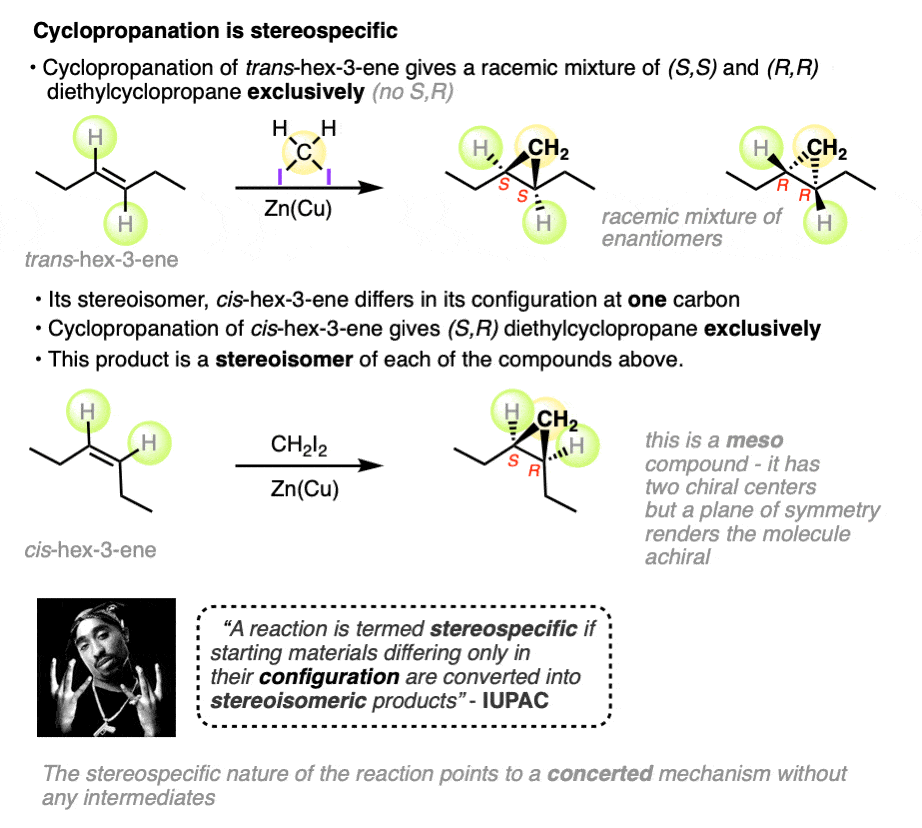
- Cyclopropanation of cis-hex-3-ene results in a single product, (3S,4R)-diethylcyclopropane (a meso compound).
- Cyclopropanation of trans-hex-3-ene results in a racemic mixture of two products, (3S, 4S)-diethylcyclopropane and (3R, 4R)-diethylcyclopropane.
The products of these reactions are stereoisomers of each other, which fulfills IUPAC’s definition of a stereospecific process. (Other examples of stereospecific reactions are halogenation of alkenes, dihydroxylation of alkenes, and epoxidation of alkenes – See article – Stereoselective and Stereospecific Reactions).
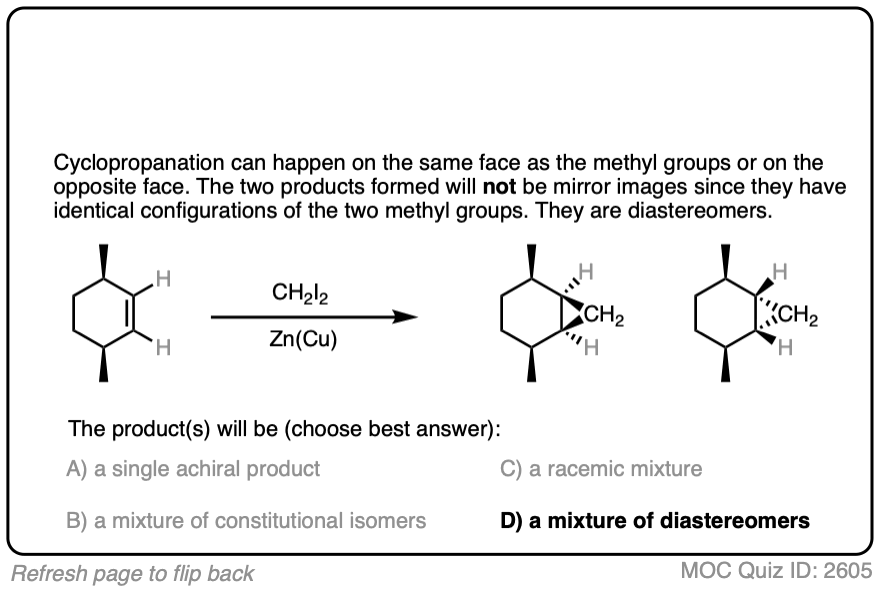
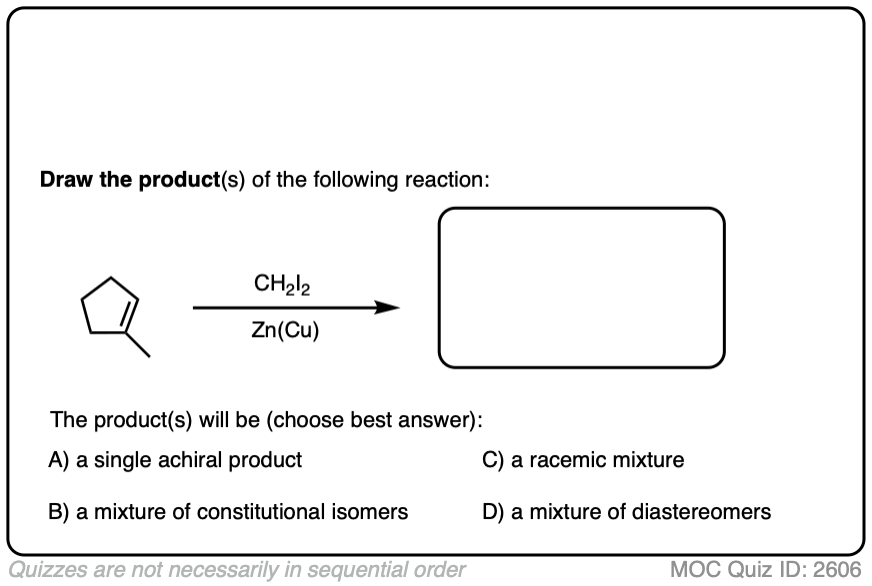
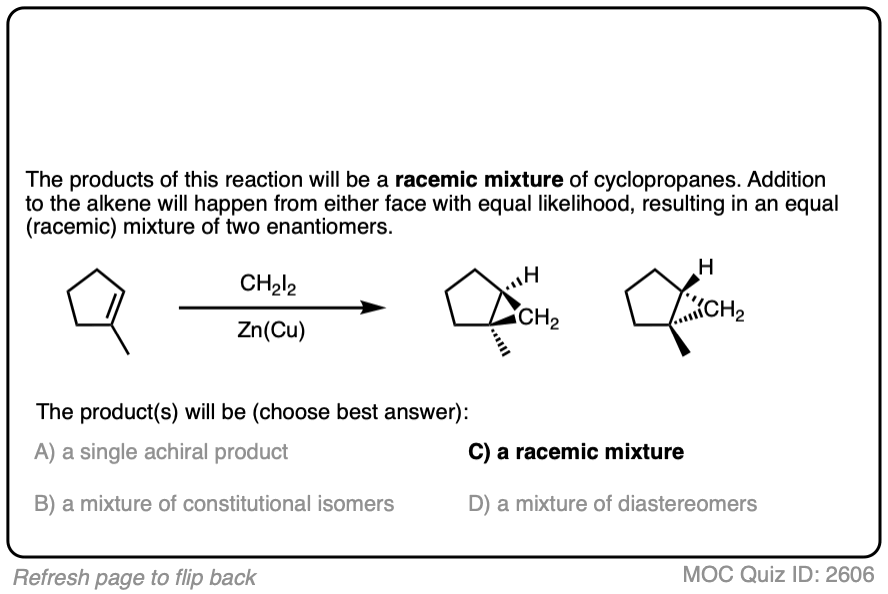
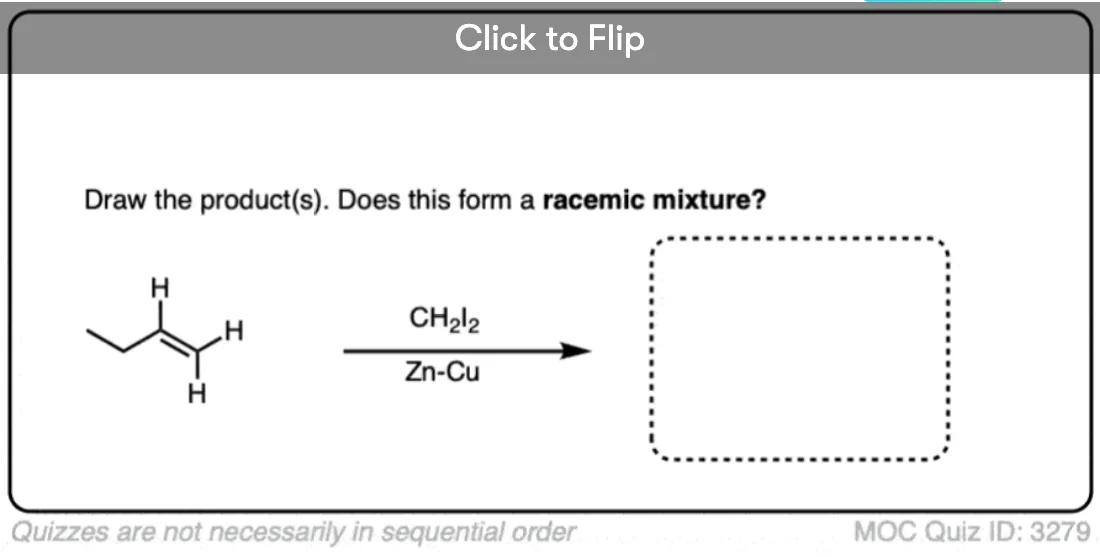
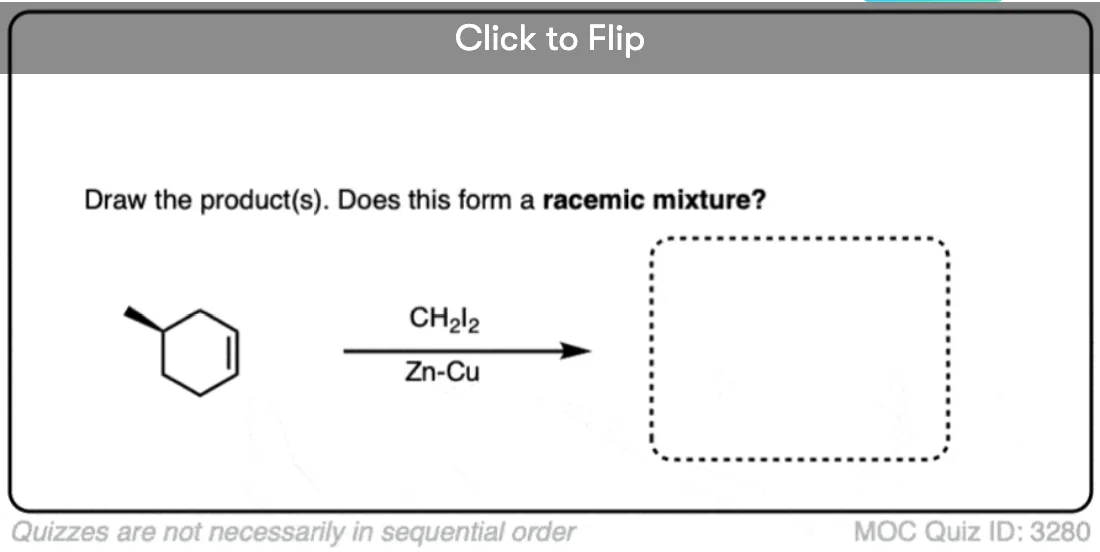
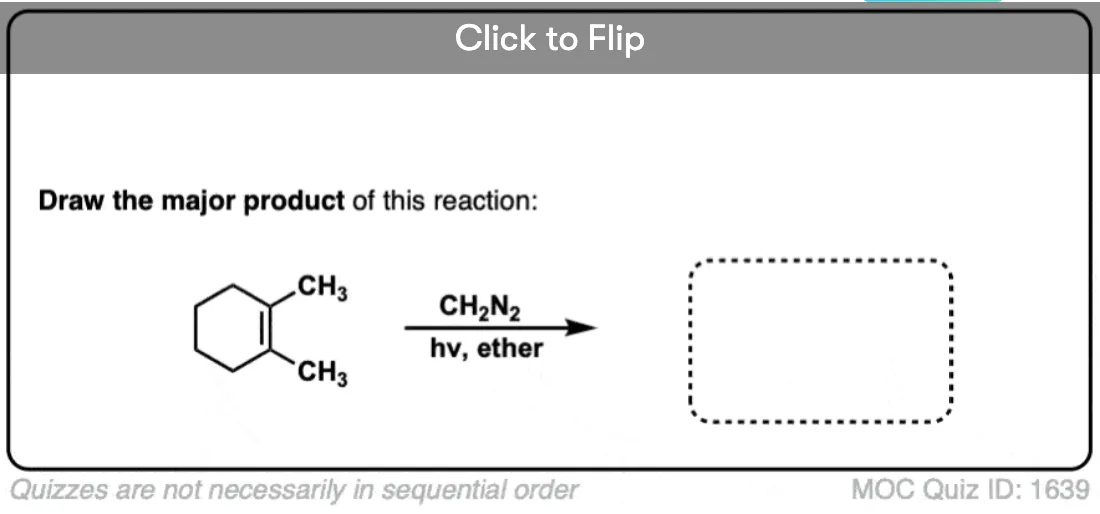
See if you can draw the product of the reaction below and predict the relationship between the products.
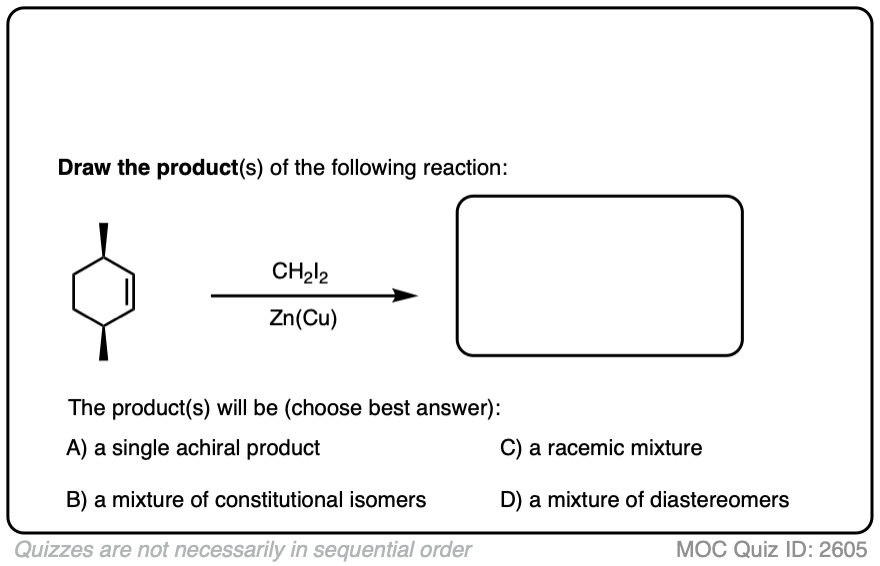 Click to Flip
Click to Flip
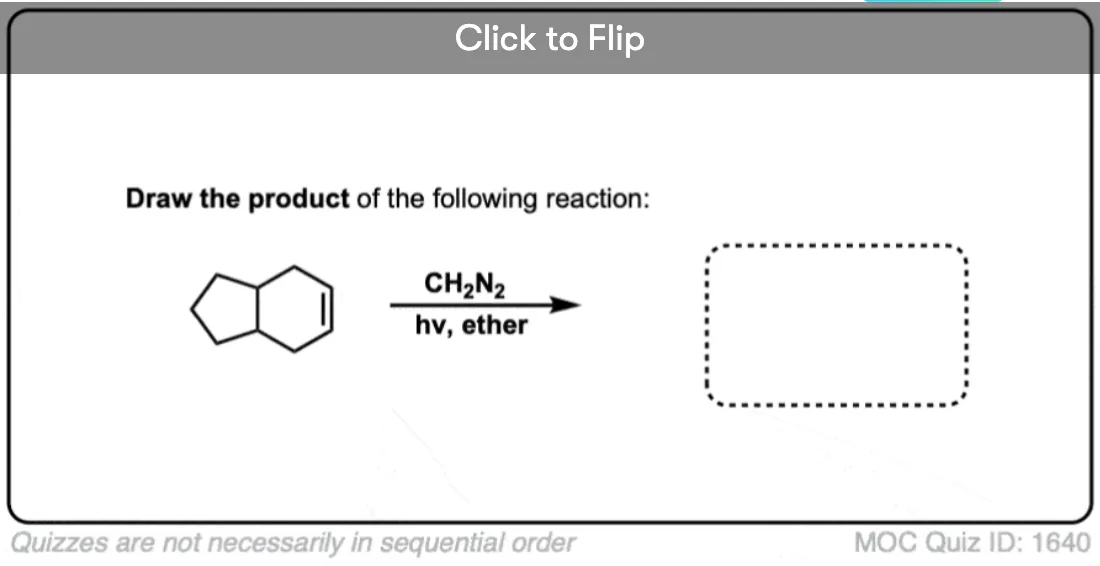
Here’s another one:
 Click to Flip
Click to Flip
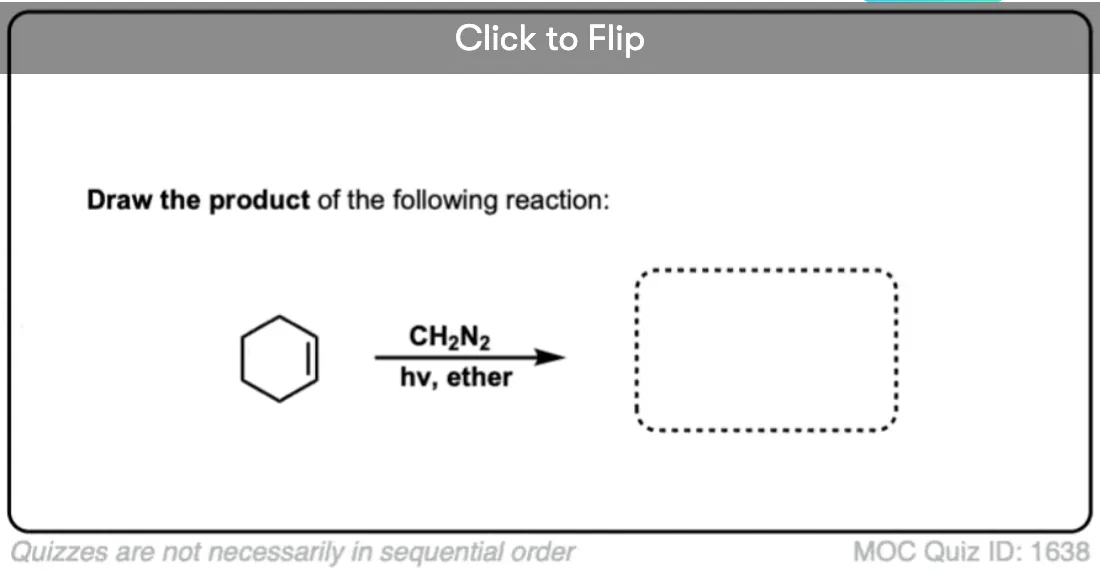
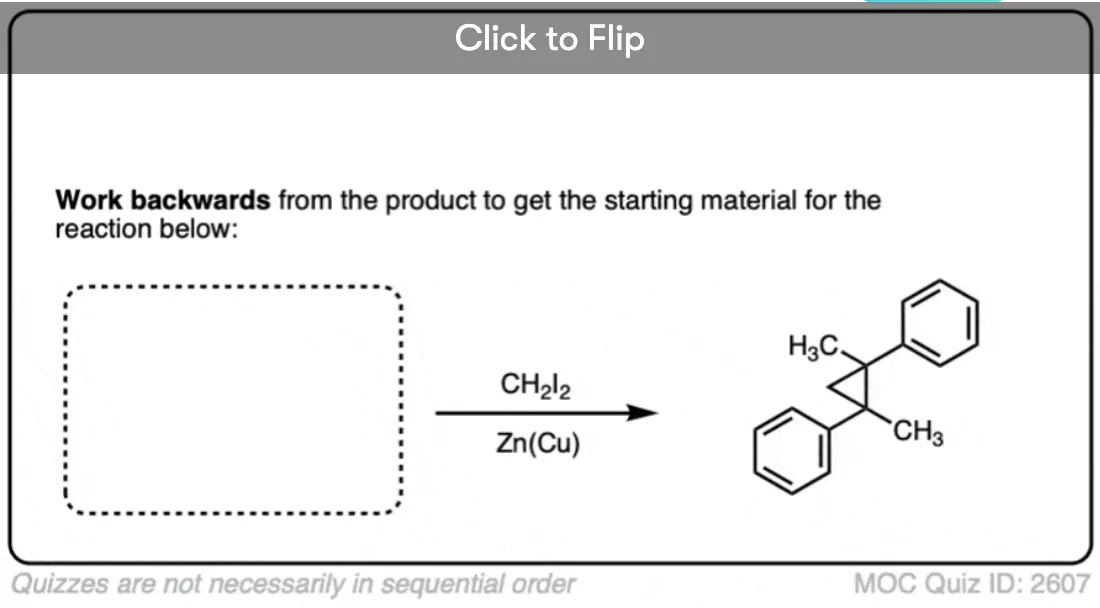
See if you can work backwards from the product below to give the starting alkene.
3. The Cyclopropanation Mechanism – Photolysis of CH2N2
Now that we’ve seen a few examples of cyclopropanation, the next logical question is to ask how it works.
In introductory courses it’s somewhat rare to go into much detail on this. One reason is that the cyclopropane products don’t really participate in many reactions we subsequently cover in this course, so given the inevitable time constraints, their mechanisms get glossed over.
We’ll cover it briefly because it provides a fun excursion into the world of carbenes.
When diazomethane is treated with light, the C-N bond breaks to liberate nitrogen gas (N2). (This is called, “photolysis”, literally, “breaking with light”).
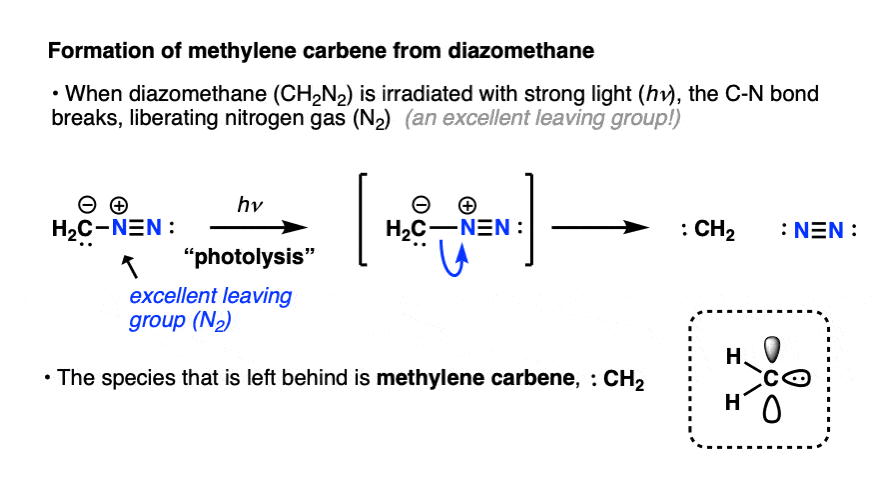
So what’s left behind?
If you follow the curved arrow, you’ll be led to the conclusion that the species left behind is simply a CH2 group with a lone pair on it. This is known as methylene carbene. Carbenes, like carbocations and free radicals, are highly reactive, electron-deficient reaction intermediates.
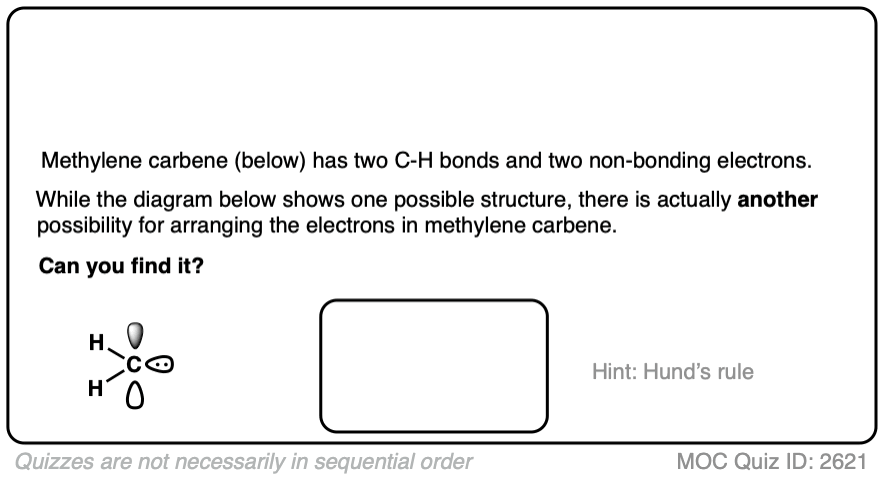
Since we’re here, why not take a quick quiz. What would you expect to be the 1) formal charge and 2) the hybridization of the central carbon in methylene carbene?
 Click to Flip
Click to Flip

Methylene carbene is a neutral, six-electron species. It is extremely reactive and pretty much impossible to control. Of all the wild horses in our stable of organic chemistry reagents, I can’t think of a wilder one than methylene carbene.
For best results, the alkene should be used as solvent. Even solvents we consider relatively inert, like pentane and benzene, will undergo reaction with methylene carbene. (For some of methylene carbene’s greatest hits, see this Note 1)
As we saw earlier, the cyclopropanation of alkenes is stereospecific. This is a clue that the reaction proceeds through a concerted mechanism.
(Reactions that aren’t stereospecific, such as addition of H-X to alkenes, and free-radical addition of H-Br to alkenes, tend to proceed through carbocation or free-radical intermediates that can undergo bond rotation.)
The mechanism for the reaction can be drawn as the concerted addition of methylene carbene to the C-C pi bond as in the drawing below.
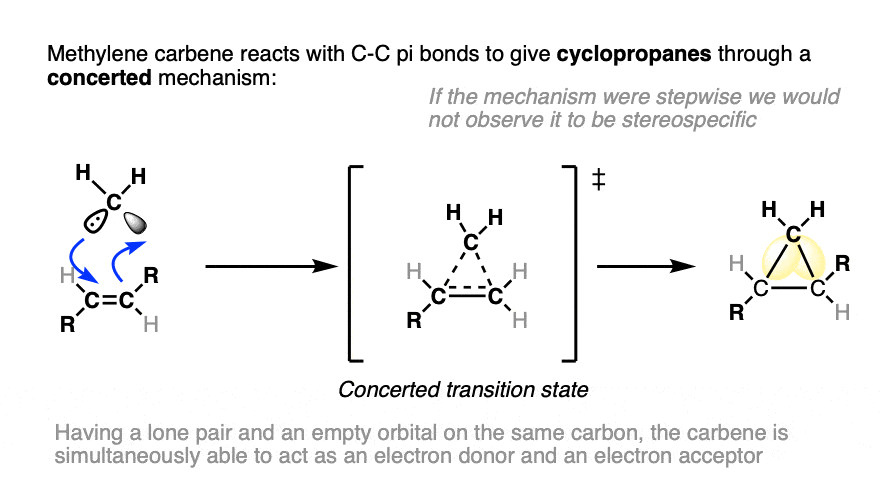
For the purposes of arrow pushing, you can think of the carbene as possessing both nucleophilic and electrophilic character; the lone pair on carbon forms one of the C-C bonds, whereas the empty orbital on the carbene carbon accepts a pair of electrons from the alkene pi bond.
There is actually another way to distribute the electrons in methylene carbene that would result in a stepwise mechanism. See Note 2]
4. The Simmons-Smith Cyclopropanation Mechanism
Cyclopropanation through the Simmons-Smith method, CH2I2/Zn(Cu), also passes through a concerted transition state.
Again, if you’re taking an introductory course, this mechanism is rarely tested, so think of this as a fun bonus excursion.
It might be helpful to think of the reaction between CH2I2 and zinc-copper couple as a fancy version of the Grignard reaction (See article – Formation of Grignard and Organolithium Reagents). The carbon-halogen bond breaks, and the metal atom (zinc in this case) inserts itself between the carbon and the halogen.
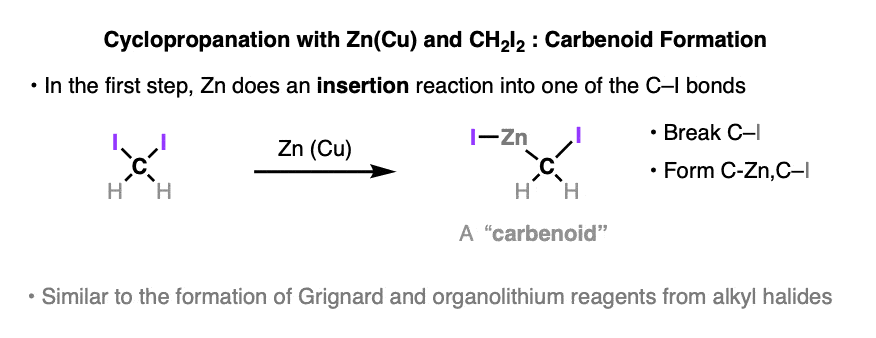
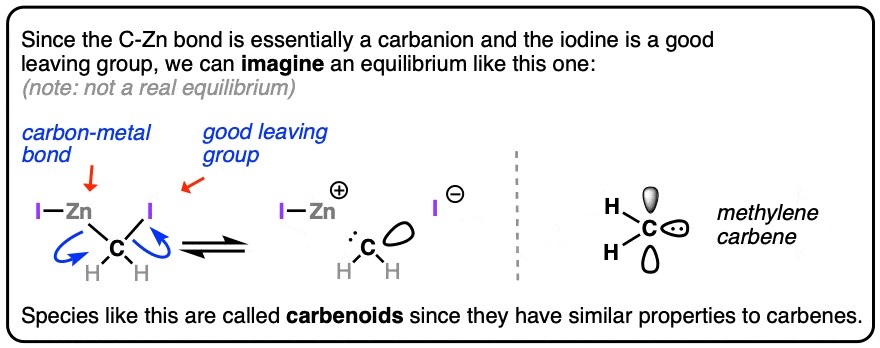
Since carbon is much more electronegative (2.5) than zinc (1.65), the carbon bonded to zinc will have significant negative charge and behave much like a carbanion. And since the carbon is also bonded to iodine, a good leaving group, it’s not too far off to imagine an equilibrium where I-Zn-CH2-I loses I-Zn(+) and I(-) , giving methylene carbene.
For that reason these types of species are known as carbenoids. To see an image of this “equilibrium” , hover here or click this link.
{kind=link}
Think of them as kinder, gentler versions of methylene carbene. They don’t react with solvents like diethyl ether, for instance.
The proposed concerted mechanism for the Simmons Smith cyclopropanation is similar to that for methylene carbene, except it incorporates loss of the carbon-zinc and the carbon-iodine bonds. [Ref]
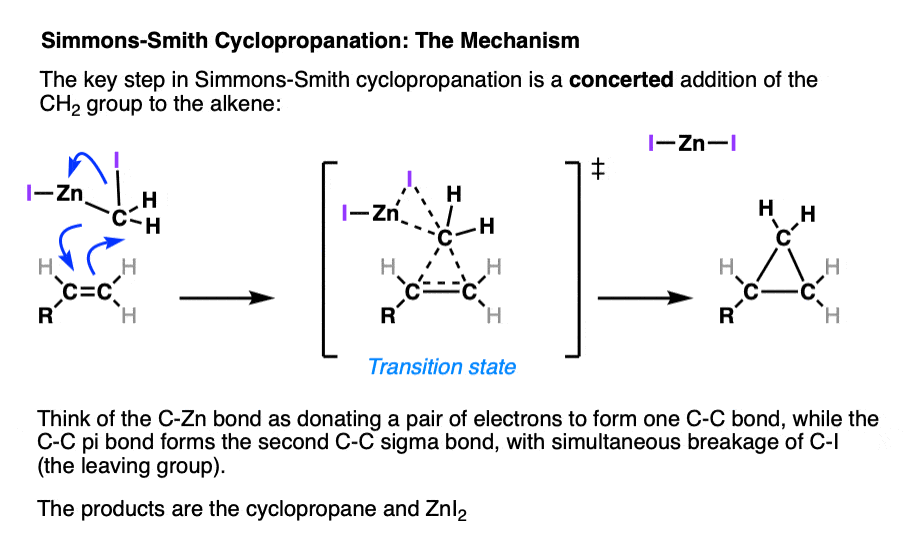
Note that the Simmons-Smith cyclopropanation only really works for CH2I2 . An attempt to use diiodoethane (CH3CHI2) for cyclopropanation gave a very low yield (4%). [Note 3]
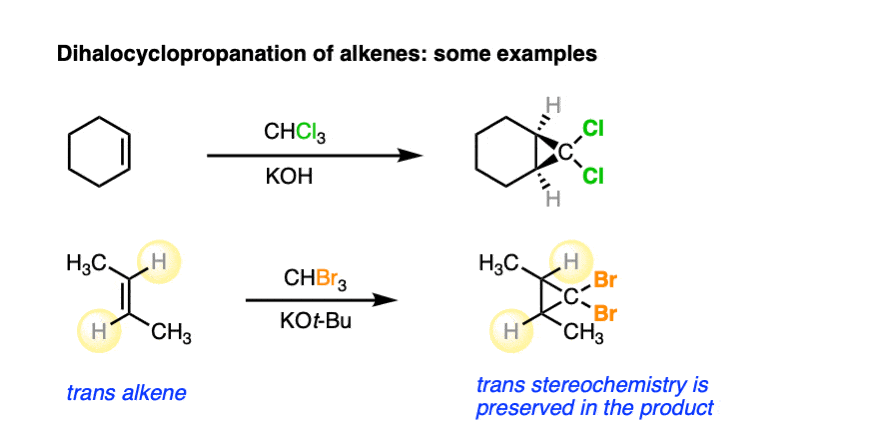
5. Dihalocyclopropanation of Alkenes
Another kinder, gentler procedure for the cyclopropanation of alkenes involves the treatment of haloforms (e.g. CHCl3, CHBr3, CHI3) with strong base.
For example, when an alkene is treated with bromoform (CHBr3) in the presence of the strong base potassium t-butoxide, the dihalocyclopropane is formed.
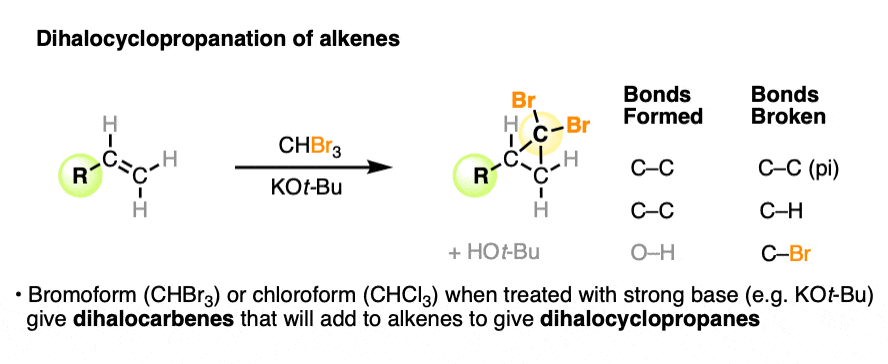
The reaction works well with chloroform and bromoform. (It presumably would work well with iodoform too, but C-I bonds are very sensitive to light.)
Like the cyclopropanation reactions we’ve seen previously, the reaction is also stereospecific.
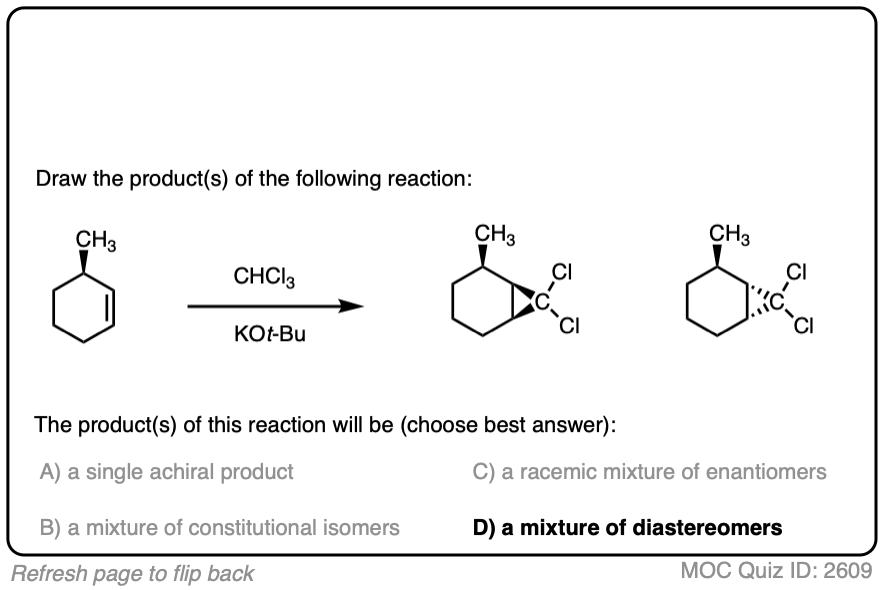
See if you can predict the products:
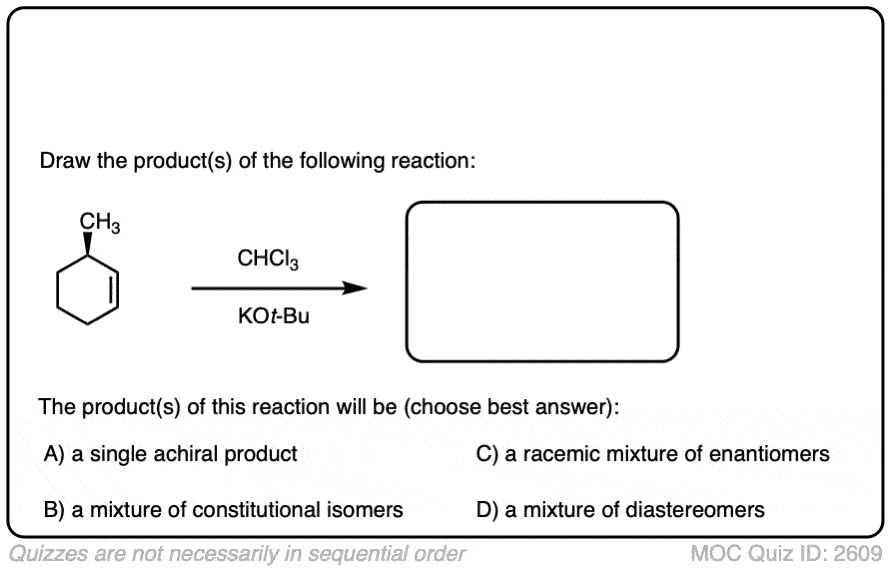 Click to Flip
Click to Flip
Another example:
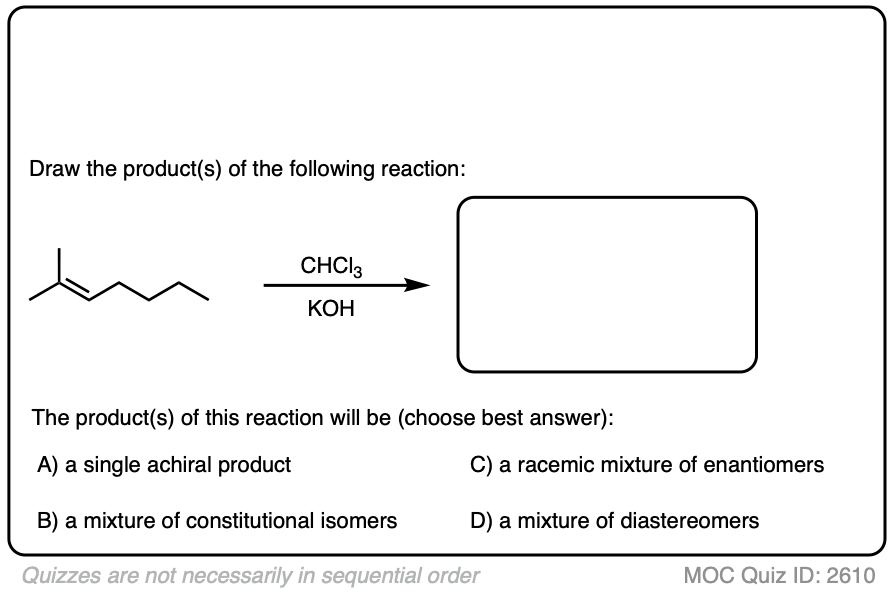 Click to Flip
Click to Flip
6. Dihalocyclopropanation – The Mechanism
So what’s going on here? Since no reaction happens in the absence of base, we should expect that the first step is deprotonation of whatever species is the strongest acid in solution. That would be the haloform.
Although bases like hydroxide and t-butoxide don’t normally deprotonate C-H bonds, the presence of three electronegative C-halogen bonds helps to make the C-H bond of CHCl3 and CHBr3 considerably more acidic than a normal C-H bond. The estimated pKa of chloroform is about 15.7, very similar to that of alcohols and water. [Ref]
Once the haloform is deprotonated to give a carbanion, it can subsequently undergo loss of a leaving group to give the neutral dihalocarbene.
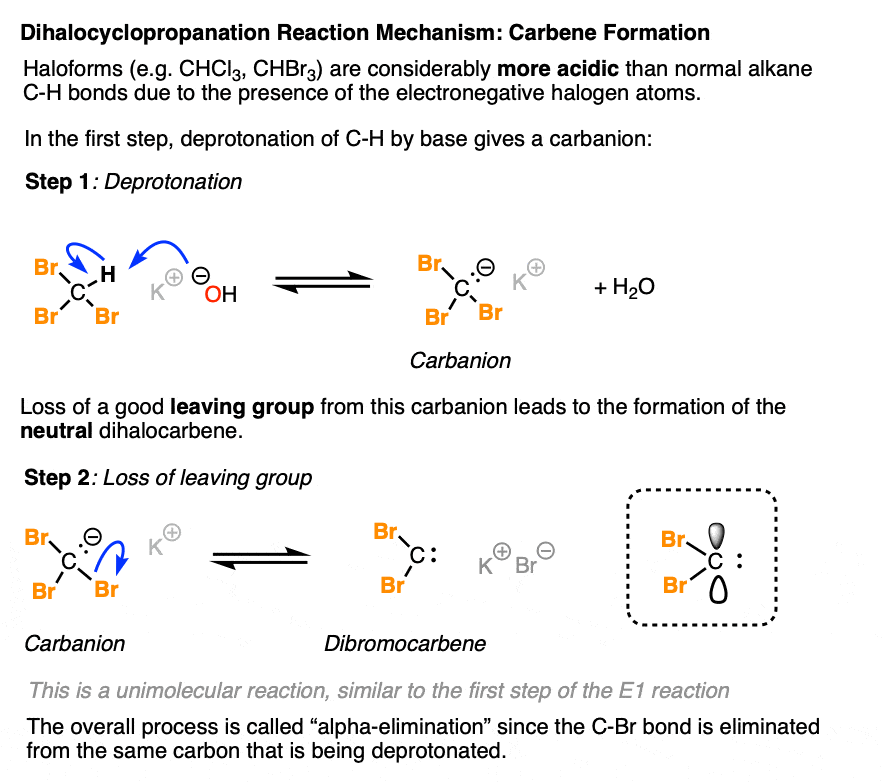
This overall process – breakage of C-H and loss of a leaving group on the same carbon – is known as alpha-elimination, as distinguished from beta-elimination where the leaving group departs from the carbon adjacent to the carbon being deprotonated (e.g. in the E1 and E2 mechanisms).
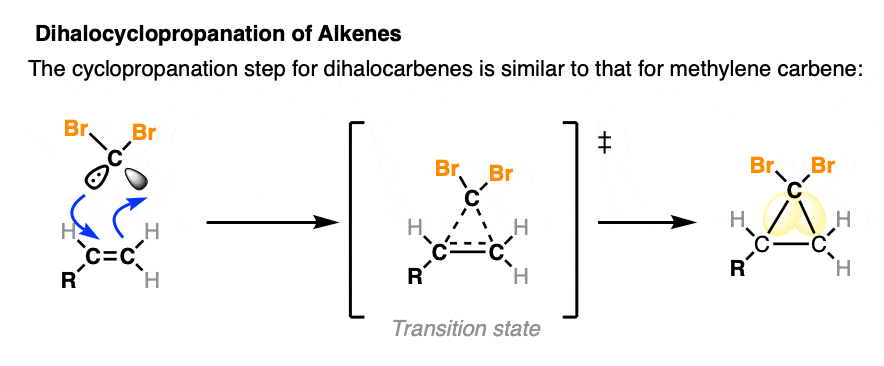
Once the dihalocarbene is formed, the next step is cyclopropanation of the alkene, which proceeds similarly to what we’ve seen earlier:
7. Reactions of Cyclopropanes (?)
Having considerable ring strain (27 kcal/mol) cyclopropanes are somewhat spring-loaded towards reactions that will result in opening the ring. They are similar to epoxides in this regard (See article: Ring Opening of Epoxides).
However, unlike epoxides there aren’t really any general reactions of cyclopropanes that are typically covered in introductory organic chemistry. The main reason is that the leaving group is carbon instead of oxygen, and carbanions are much stronger bases than alkoxides and therefore worse leaving groups. Opening a cyclopropane with a nucleophile is difficult unless you have a good electron withdrawing group attached to it that can delocalize the charge.
So for the purposes of introductory organic chemistry, cyclopropane formation is one of those reactions that is presented in the chapter on alkenes but doesn’t really get developed further at a later part in the course. For that reason it’s often the first reaction to be dropped when instructors are short on time.
8. Summary
Alkenes combine with carbenes (and carbenoids) to give cyclopropanes. We saw a variety of ways of making carbenes.
Cyclopropanation of alkenes is similar to dihydroxylation, epoxidation, and hydrogenation of alkenes in that
- The reaction proceeds through a concerted transition state
- The reaction gives the products of syn addition
- The reaction is stereospecific
For studying purposes, I generally suggest grouping these reactions together with hydroboration to give a family of reactions some may reasonably call “The Concerted Pathway” (See article: Alkene Addition Pattern #3, The “Concerted Pathway“).
Notes
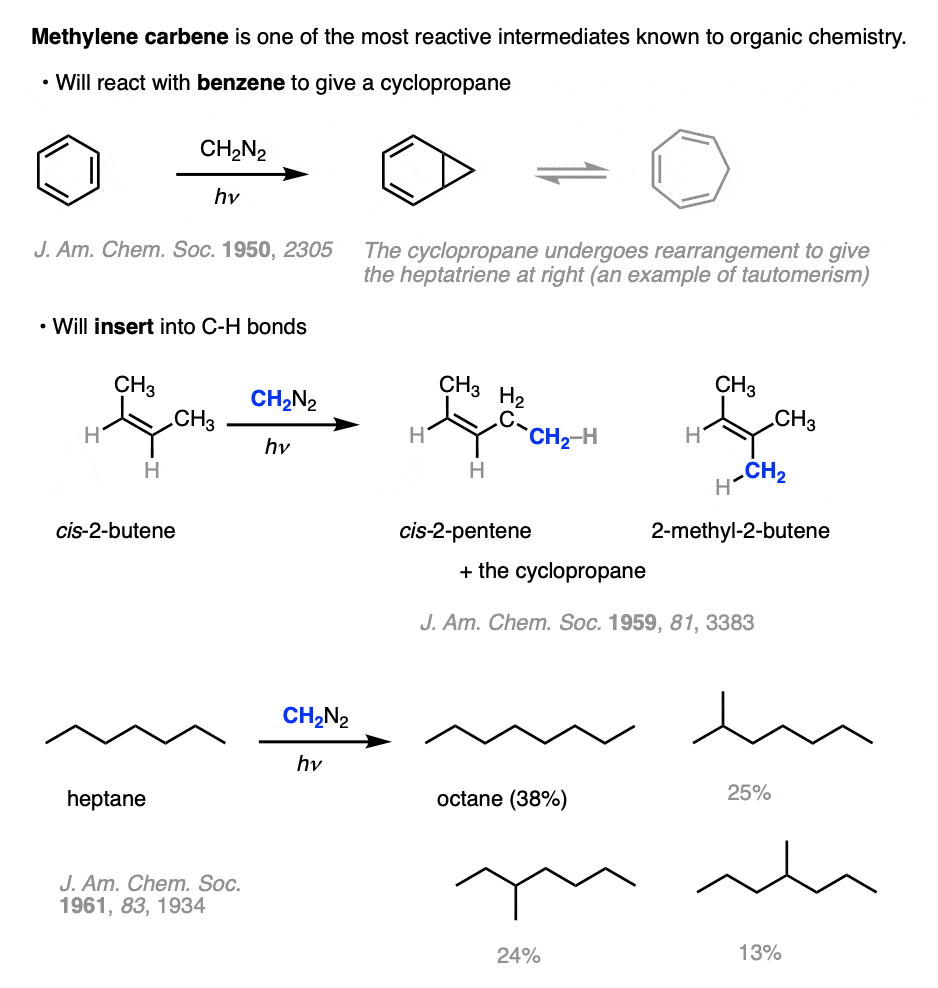
Note 1. Methylene carbene will do things that few other reactive intermediates will do, such as participating in reactions with relatively inert compounds like benzene and alkanes. The problem is that it is impossible to control.
For example, it will make cyclopropanes from benzene.
The other amazing reaction methylene carbene will do is to insert itself into C-H bonds, resulting in the lengthening of alkanes (and alkenes) by a single CH2 unit.
Two of the side products in this cyclopropanation study were cis-2-pentene and 2-methyl-2-butene.
Methylene will react with ordinary alkanes as well. In heptane, it gives a mixture of n-octane and octane isomers. The reaction is not very selective, however.
Note 2. There is actually another way to draw the electron configuration of a carbene.
 Click to Flip
Click to Flip
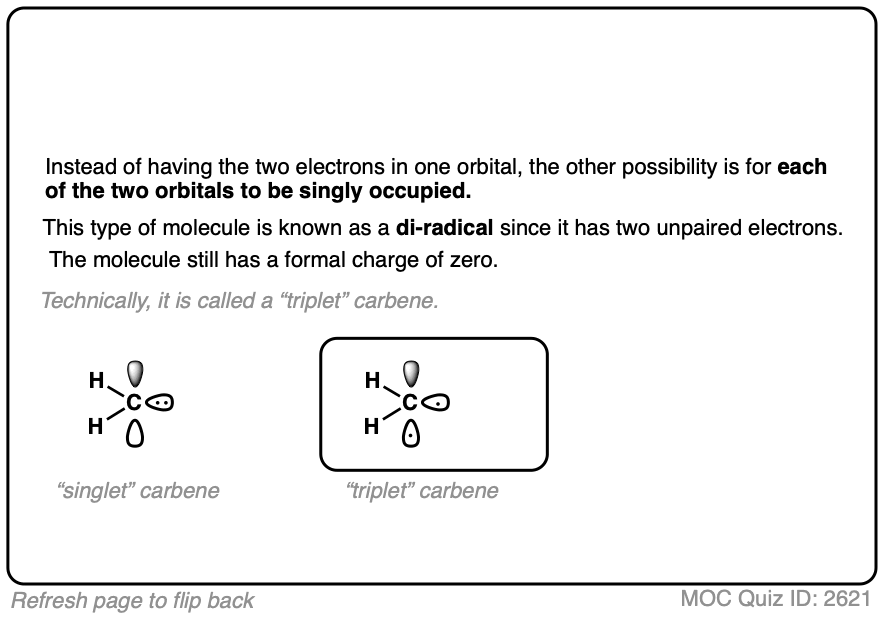
One of the important conclusions of the 1959 Snell study [Ref] was that the stereospecificity of the reaction showed that the reaction essentially went through a singlet carbene. Had cyclopropanation gone through addition of a triplet carbene, there would be an intermediate carbon radical capable of undergoing bond rotation, which would have led to a mixture of stereoisomers.
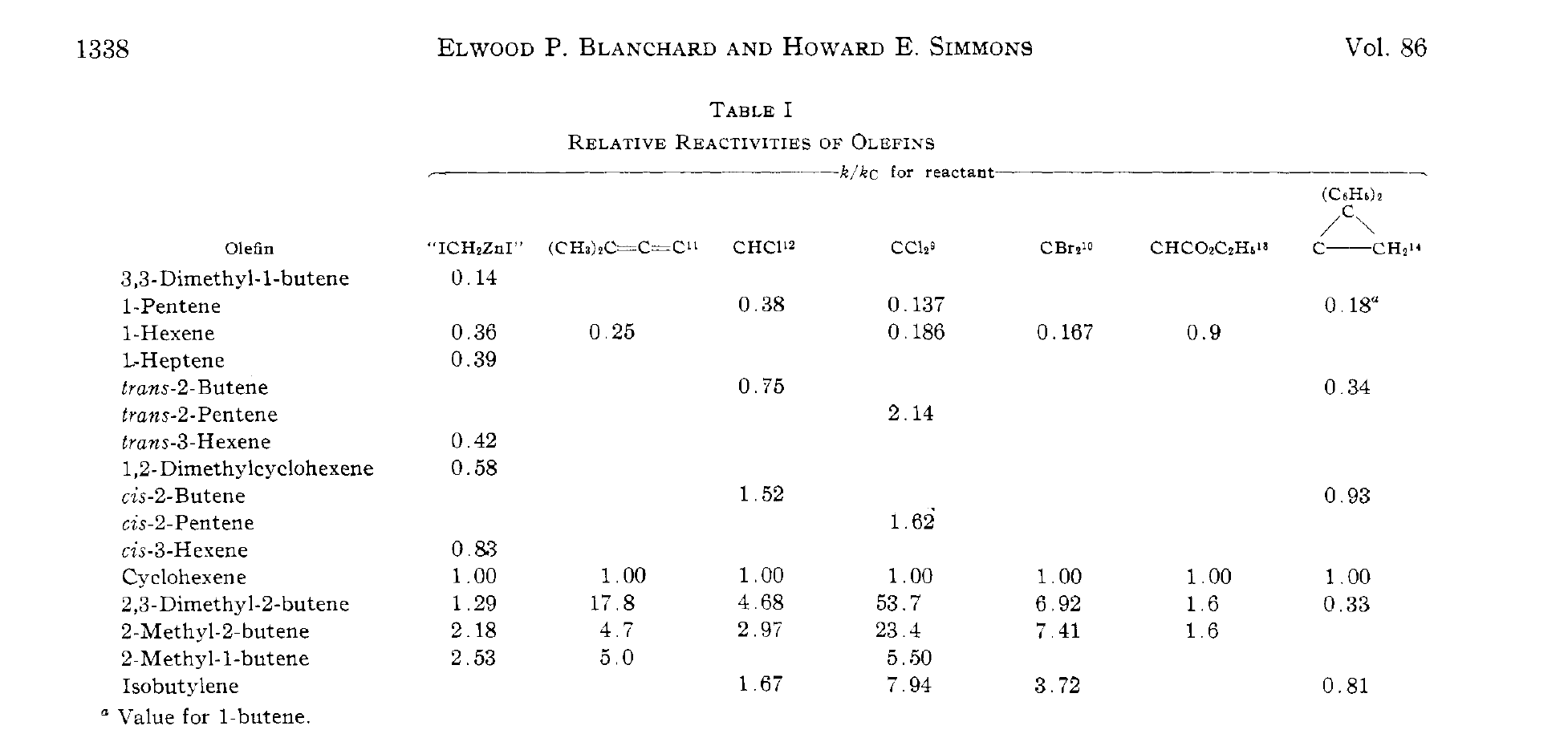
Note 3. Scope of the Simmons Smith cyclopropanation process [Ref] (from 1959 – many modifications have been made since then!)
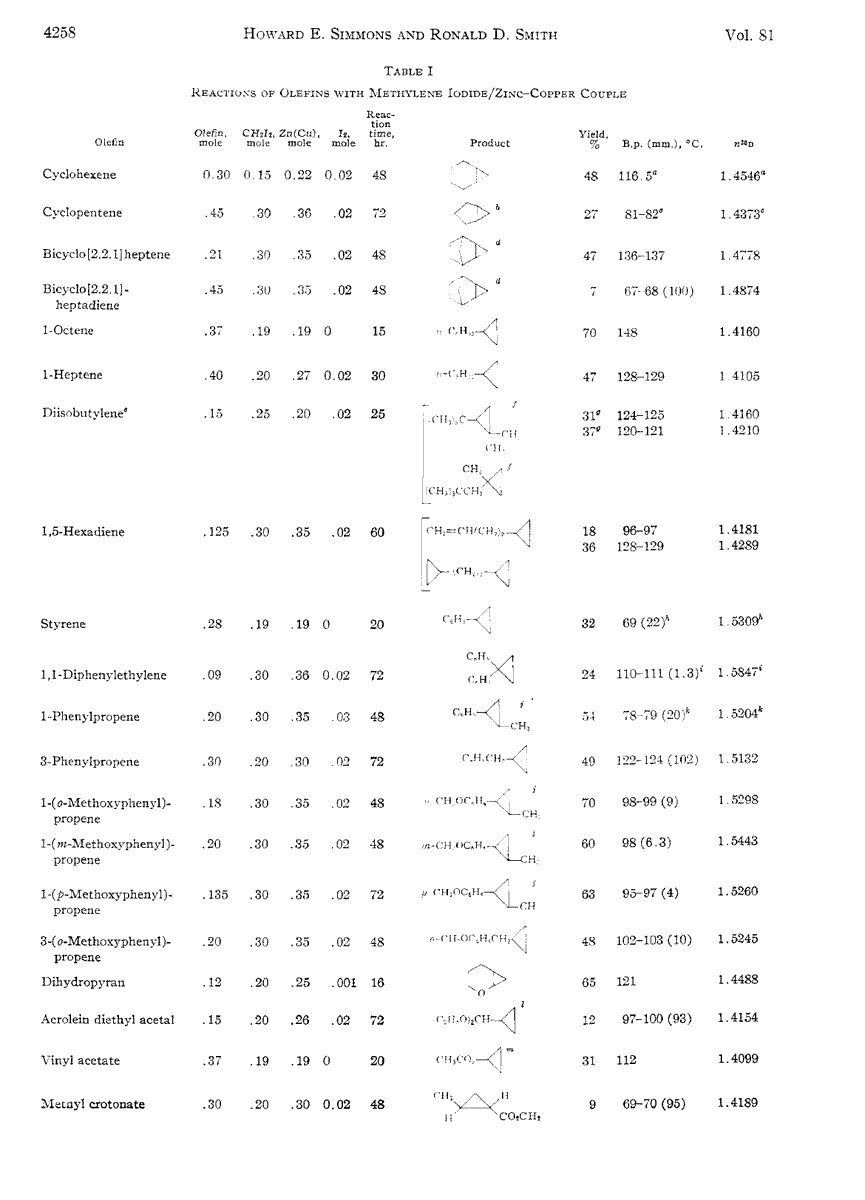
Generally, the more electron rich the alkene, the more reactive it is with the Simmons-Smith reagent, although steric hindrance can play a role.
(This is from JACS 1964, 1347)
Note 4. Cyclopropanation of alkynes is also possible. This naturally occurring cyclopropene was formed from the cyclopropanation of an alkyne, a mere 2 years after the original Simmons-Smith report (albeit in 4% yield)

Note 5. Many natural products contain cyclopropanes. The current record is 6, contained in this molecule known as U-106305, isolated in 1990 from fermentation of a Streptomyces variant.
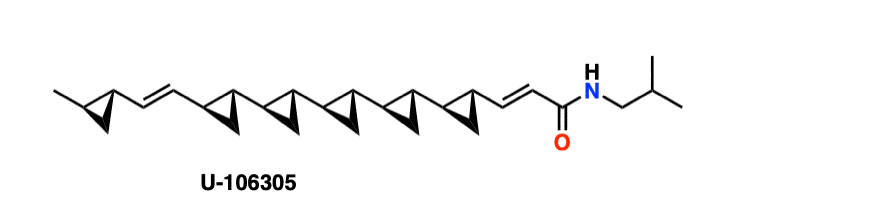
It has been synthesized (Ref) by the research group of Prof. Andre Charette (U. Montreal) through application of successive Simmons-Smith cyclopropanation reactions. Charette’s research group has made many advances in the synthesis of enantiomerically enriched cyclopropanes.
Quiz Yourself!
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading
Simmons-Smith Reaction:
- A New Synthesis of Cyclopropanes
Howard E. Simmons and Ronald D. Smith
Journal of the American Chemical Society 1959 81 (16), 4256-4264
DOI: 10.1021/ja01525a036
This full paper, expanded from the original report (JACS 1958 5323) explains the development of the reaction and provides a lot of experimental detail on its scope (broad), its stereospecificity, and proposes a mechanism. - Cyclopropanes from Unsaturated Compounds, Methylene Iodide, and Zinc-Copper Couple
Simmons, H. E.; Cairns, T. L.; Vladuchick, S. A.; Hoiness, S. A. Org. Reactions 1973, 20, 1-133
DOI: 10.1002/0471264180.or020.01
This chapter in Organic Reactions has everything you need to know about the Simmons-Smith reaction, including mechanistic studies, experimental procedures, and substrate scope. - NORCARANE
D. Smith and H. E. Simmons
Org. Synth. 1961, 41, 72
DOI: 10.15227/orgsyn.041.0072
A detailed procedure for the Simmons-Smith reaction in Organic Syntheses, including the preparation of the Zn-Cu couple. - The Addition of Dichlorocarbene to Olefins
William von E. Doering and A. Kentaro Hoffmann
Journal of the American Chemical Society 1954 76 (23), 6162-6165
DOI: 10.1021/ja01652a087
The original paper describing the synthesis of dichlorocyclopropanes from alkenes with chloroform and KOtBu, by William von Eggers Doering. - 1,6-Methano[10]Annulene
E. Vogel, W. Klug, and A. Bruer
Org. Synth. 1974 54, 11
DOI: 10.15227/orgsyn.054.0011
The second step in this procedure is the synthesis of a dichlorocyclopropane, by addition of dichlorocarbene to an alkene. - Methylene, CH2. Stereospecific Reaction with cis- and trans-2-Butene
Robert C. Woodworth and Philip S. Skell
Journal of the American Chemical Society 1959 81 (13), 3383-3386
DOI: 10.1021/ja01522a058
In this study the authors find that photolysis of diazomethane in the presence of cis– and trans– 2-butene results in stereospecific cyclopropanation, indicating that it goes through a single carbene intermediate. - A DFT Study of the Simmons−Smith Cyclopropanation Reaction
Fernando Bernardi, Andrea Bottoni, and Gian Pietro Miscione
Journal of the American Chemical Society 1997 119 (50), 12300-12305
DOI: 10.1021/ja971995x
Calculations of the transition state for cyclopropanation of alkenes. - Stereoselective Cyclopropanation Reactions
Hélène Lebel, Jean-François Marcoux, Carmela Molinaro, and André B. Charette
Chemical Reviews 2003 103 (4), 977-1050
DOI: 10.1021/cr010007e
Since the development of the Simmons-Smith reaction a tremendous amount of work has gone into developing enantioselective variants. This review is quite comprehensive. - SYNTHESIS OF STERCULIC ACID
Nicholas T. Castellucci and Claibourne E. GriffinJournal of the American Chemical Society 1960 82 (15), 4107-4107
DOI: 10.1021/ja01500a070
Synthesis of a naturally occurring cyclopropene through cyclopropanation of an alkyne. - Cyclopropane Synthesis from Methylene Iodide, Zinc-Copper Couple, and Olefins. III. The Methylene-Transfer Reaction
Howard E. Simmons, Elwood P. Blanchard, and Ronald D. Smith
Journal of the American Chemical Society 1964 86 (7), 1347-1356
DOI: 10.1021/ja01061a017
Further study on the Simmons-Smith reaction, studying the mechanism and properties of the active reagent.
Thank You for your work. I think that there might be a mistake in section three – 3. The Cyclopropanation Mechanism – Photolysis of CH2N2. The second picture(the last in the section) shown no second “R” on the alkene in the concerted transition state, the “R” appears just after that, so it made me think that there is a mistake.
Oh snap, you are correct. I can fix this. Thank you so much and please let me know if you see anything else?
I might come off as a bit impatient, but when will you put in the notes? I am very excited to read them.
This week. I’m happy to learn that at least one person reads through to the notes at the end!