Reactions of Aromatic Molecules
Electrophilic Aromatic Substitution: Introduction
Last updated: May 28th, 2026 |
The Six Key Electrophilic Aromatic Substitution Reactions
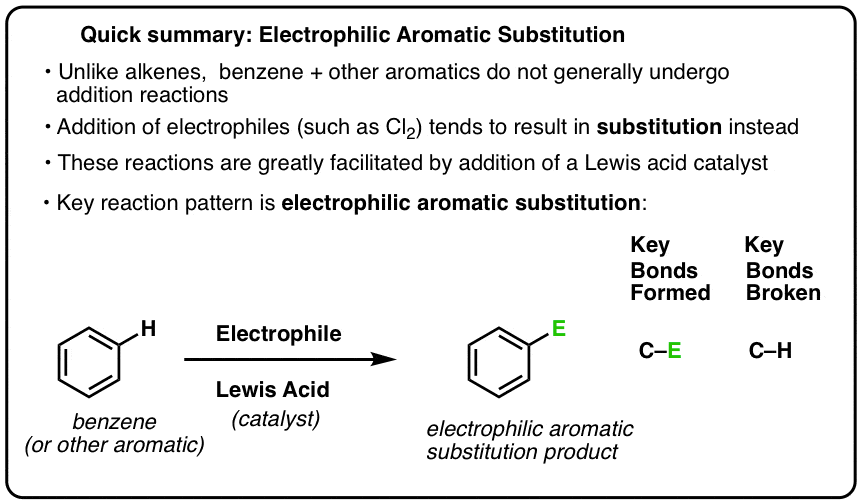
- In electrophilic aromatic substitution a C-H bond is broken and a new C-E bond (E being an electrophilic atom such as Cl, Br, N…) is formed.
- There are six key electrophilic aromatic substitution reactions in most introductory organic chemistry courses: chlorination, bromination, nitration, sulfonation, Friedel-Crafts alkylation, and Friedel-Crafts acylation.
- Each of these reactions requires an acid catalyst to activate it so that the relatively unreactive aromatic ring will attack it.
{kind=link}
- Alkenes Give “Addition” Products Upon Reaction With Electrophiles. So How Does Benzene Compare?
- Electrophilic Aromatic Substitution
- Lewis Acids Accelerate The Rate of Electrophilic Aromatic Substitution Reactions
- The Key Pattern For Six Important Electrophilic Aromatic Substitution Reactions
- Aromatic Chlorination and Bromination
- Aromatic Nitration and Sulfonation
- Friedel-Crafts Alkylation and Acylation
- Summary: The Six Key Electrophilic Aromatic Substitution Reactions
- Notes
- Quiz Yourself!
1. Alkenes Give “Addition” Products Upon Reaction With Electrophiles. So How Does Benzene Compare?
When we covered the reactions of alkenes a while back – a lot of reactions! – we saw that the vast majority fell into the class of reactions we call addition reactions. That’s where we break a (relatively weak) C-C (pi) bond and form two new single bonds to carbon. Chlorination of alkenes with Cl2 is a classic example:
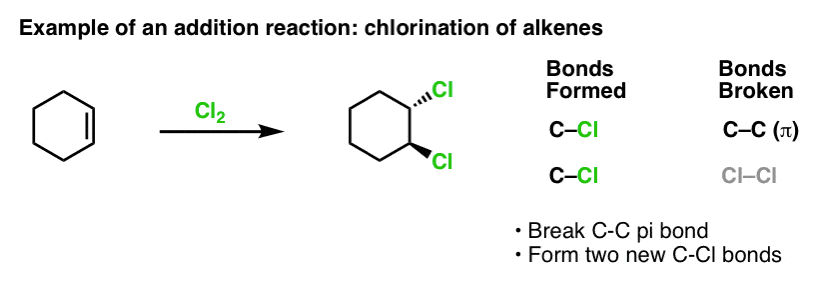
Since we’re on the topic of benzene, it’s natural to wonder how well the pi bonds in aromatic systems (like benzene) compare in reactivity to the pi bonds in alkenes, and by extension, how the reactions of aromatic compounds compare to the reactions of alkenes.
We’ve seen that benzene has unusual stability (36 kcal/mol of resonance energy) relative to what we’d expect for theoretical “cyclohexatriene”, which would certainly lead us to predict that the pi bonds in aromatic molecules will be less reactive, relative to alkenes.
You might also recall that benzene itself is unusually difficult to hydrogenate. Hydrogen (H2) can be made to add across most alkenes in the presence of a catalytic amount of finely divided palladium on carbon (Pd/C), but you really need to break out the can of Aldrich Brand Whup-Ass® (i.e. high temperatures, high pressures of H2 , extended reaction times ) in order to successfully add H2 to benzene, relative to “typical” alkenes.
{kind=link}
Knowing this, how might we expect electrophiles like Cl2 or Br2 to react with aromatic compounds like benzene?
2. Electrophilic Aromatic Substitution
Our first guess might be that benzene reacts with Cl2 to give an “addition” product like that shown below (albeit more slowly than with a “normal” alkene):
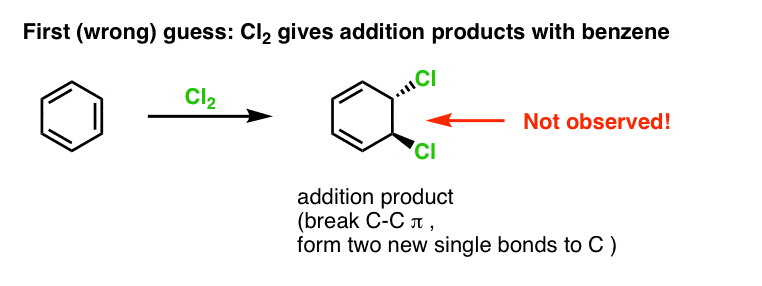
That’s actually not the product that we observe!
Instead, if you treat benzene with Cl2 you will eventually (and very slowly) obtain the following product: chlorobenzene.
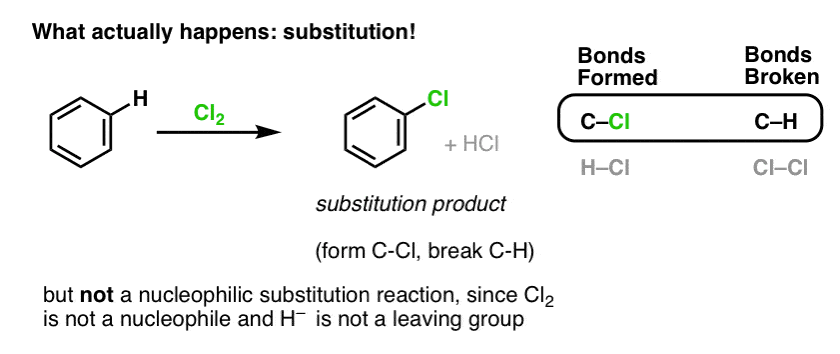
What bonds formed and what bonds broke in this reaction?
We formed C-Cl and broke C-H . Since the pi bonds are all intact, this is not an addition reaction. Instead, this is therefore a type of substitution reaction, where we form and break a bond on a single carbon.
We’ve seen an example of substitution reactions before, but those were nucleophilic substitutions, where a nucleophile (e.g. RS– ) is added to an alkyl halide electrophile (e.g. R-Br), displacing a leaving group (Br– here) forming a C-Nuc bond (C-S in this case) and breaking a C-LG bond (C-Br here).
So is the reaction of Cl2 with benzene likewise a nucleophilic substitution reaction? No!
Cl2 is an extremely poor nucleophile, reacting as an electron-donor only with strong Lewis acids (e.g. AlCl3, below). When combined with even a relatively mild nucleophile such as the pi-bond in an alkene, it behaves as an electrophile (electron acceptor) as we saw in its reaction with alkenes.
The reaction of Cl2 with benzene is thus called an electrophilic aromatic substitution (EAS for short):
- Electrophilic, because we’re adding an electron-poor species (electrophile),
- to an aromatic compound (benzene);
- substitution, because we’re breaking C-H and forming C-E, where E is our electrophile (Cl in this case).
3. Lewis Acids Accelerate The Rate of Electrophilic Aromatic Substitution Reactions
The reaction of Cl2 with benzene is faster than toenail growth, but not by much [ref] (It is, however, much faster with more electron-rich aromatics such as toluene and phenol). So instead of sitting around the lab for weeks waiting for a reaction to complete, we can add a reagent that “soups up” the reactivity of Cl2 to make it into an even better electrophile: a catalyst, in other words.
Addition of a good Lewis acid like AlCl3 or FeCl3 does the trick. [There are many other Lewis acids which will also do the job, but that’s a topic for when we get into the mechanism – not today]. update: see post on mechanism
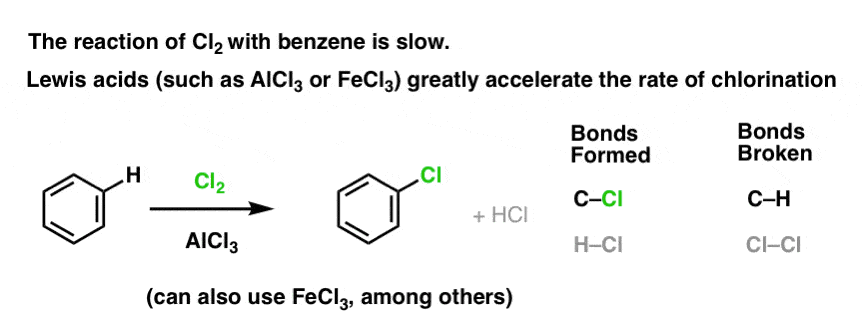
How does this work?
From our series on alcohols you may recall that alcohols (R–OH) can be coaxed to participate in substitution and elimination reactions if a strong acid is added ( forming R–OH2+ ). That’s because the conjugate acid is always a better leaving group ; H2O is a weaker base, and thus a much better leaving group, than HO– . Ethanol itself will never react with NaCl to give ethyl chloride, because the resulting leaving group HO- is too strong a base relative to Cl- for the reaction to proceed to any extent. But if we convert the alcohol into its conjugate acid R-OH2 + with a strong acid such as HCl, the reaction can then proceed, since Cl– is displacing the much weaker base H2O.
It’s can be helpful to think of H+ as weakening the C-O bond, and thus making the carbon attached to it a better electrophile.
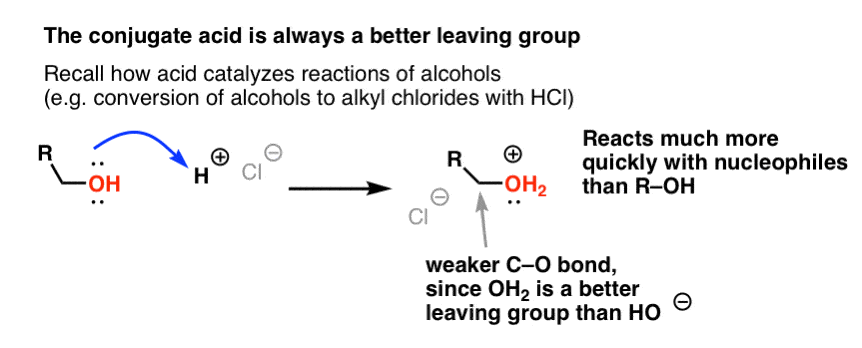
Just as acid “primes” the carbon attached to the OH group for a subsequent reaction by converting the hydroxyl group into its conjugate acid, Lewis acids similarly work the same magic on Cl2 (and for that matter, all the electrophilic aromatic substitution reactions we’ll be covering will involve some kind of acid catalysis).
In the case of chlorination, the Lewis acid (AlCl3, below) accepts a pair of electrons from Cl2. This weakens the Cl–Cl bond, making it into an even better electrophile. Attack by a nucleophile at the distal Cl will liberate not Cl- , but the even weaker base (and thus, better leaving group) AlCl4(-).
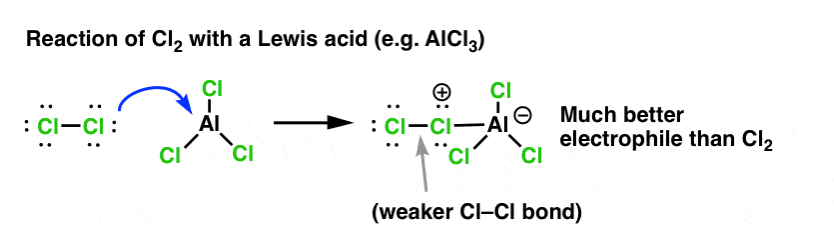
Addition of a Lewis acid (which, recall, includes Brønsted acids like H2SO4 ) is a common thread in the six key electrophilic aromatic substitutions which are generally covered in introductory organic chemistry.
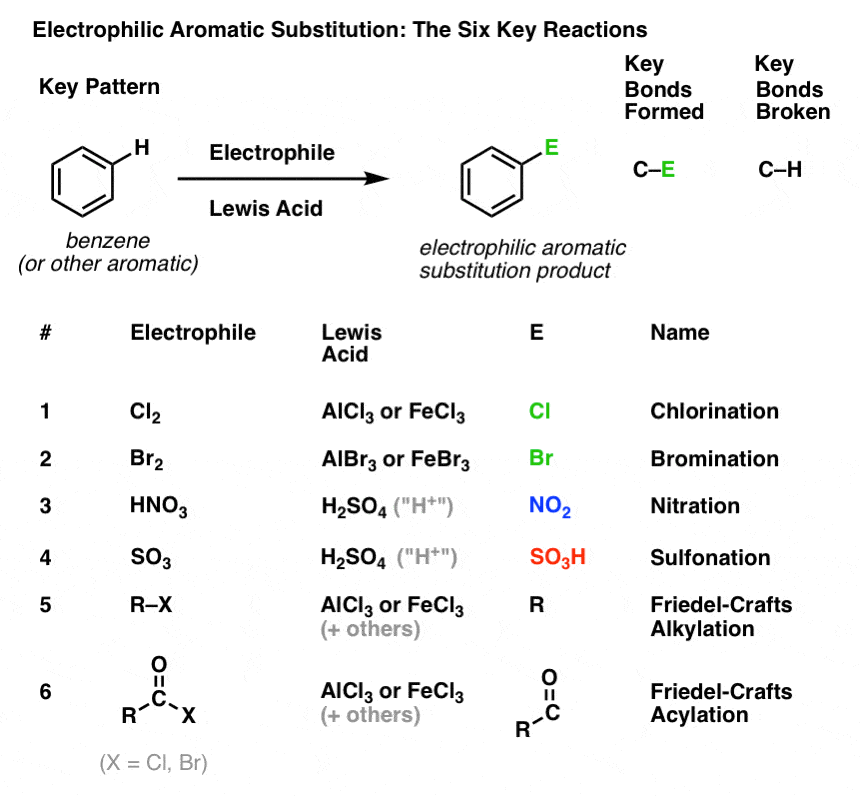
4. The Key Pattern For Six Important Electrophilic Aromatic Substitution Reactions
In this section, we’ll introduce these six key electrophilic aromatic substitution reactions, which group nicely into three pairs:
- chlorination and bromination
- nitration and sulfonylation
- Friedel-Crafts alkylation and Friedel-Crafts acylation
We’re not going to get into the mechanisms yet. The point here is just to follow what bonds form and break, so that you see the key pattern.
Here it is:
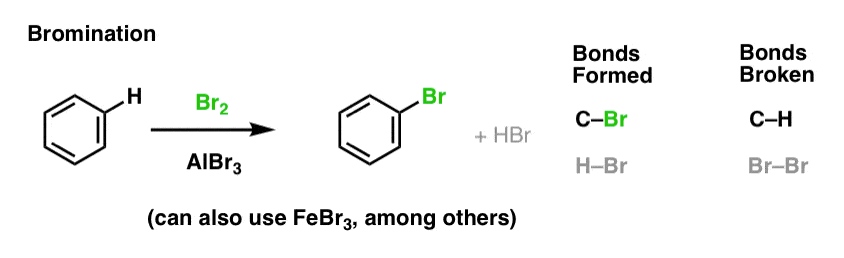
5. Chlorination and Bromination Of Aromatic Molecules
We’ve seen chlorination (above); bromination is very similar. Since Br2 by itself is not a strong enough electrophile to react with benzene at a reasonable rate, we use a Lewis acid such as AlBr3 or FeBr3 to accelerate the reaction. [Using AlBr3/FeBr3 instead of AlCl3/ FeCl3 avoids some “scrambling” of the halides, as we’ll see in a subsequent post when we get into the mechanism].
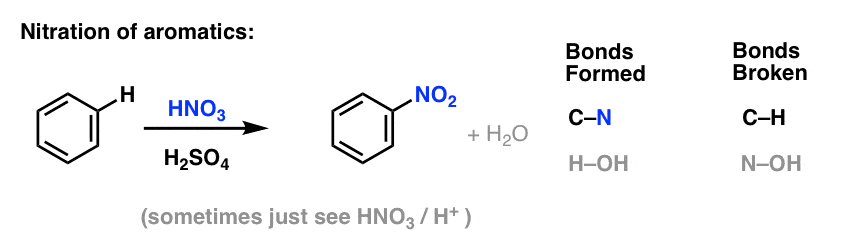
6. Nitration and Sulfonation of Aromatic Molecules
Nitration (replacement of H with a NO2 group) is not a reaction we saw in our section on alkenes, but it’s a popular reaction with aromatic molecules such as benzene. This is the reaction by which methylbenzene (toluene) is converted to 2,4,6-trinitrotoluene. You’re likely more familiar with this molecule as the high explosive TNT.
Nitration is the substitution of H with NO2, using nitric acid (HNO3) as the source of NO2 and sulphuric acid (H2SO4) as the Lewis acid:
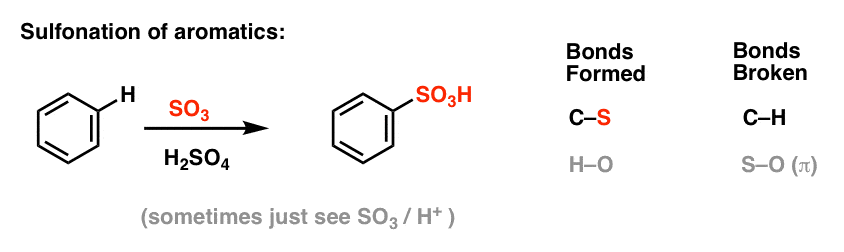
Sulfonation (replacement of H with a SO3H group) also turns out to be a useful reaction. It can be performed by adding sulfur trioxide (SO3) in the presence of sulphuric acid (H2SO4) as the Lewis acid:
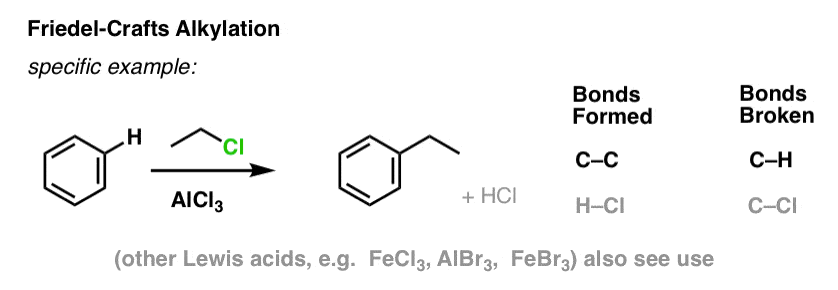
7. Friedel-Crafts Alkylation and Acylation
In the four reactions above, we observed the formation of C-Cl, C-Br, C-N and C-S.
It’s also possible to form carbon-carbon bonds to aromatic molecules by adding alkyl or acyl halides in the presence of Lewis acids. These reactions are known as Friedel-Crafts reactions, after their inventors (the reaction dates back to 1877).
In Friedel-Crafts Alkylation, we start with an alkyl halide “R-X” such as CH3CH2Cl and then add a Lewis acid such as AlCl3 or FeCl3 [there are many other Lewis acids which work, but those are the most common examples]. As with Cl2, the Lewis acid accelerates the reaction by coordinating to the halogen, weakening the C–Cl bond, and making it a better leaving group and thus allowing the attached carbon to be more easily attacked by nucleophiles. No reaction occurs without the Lewis acid.
Note that here we form C–C and break C–H.
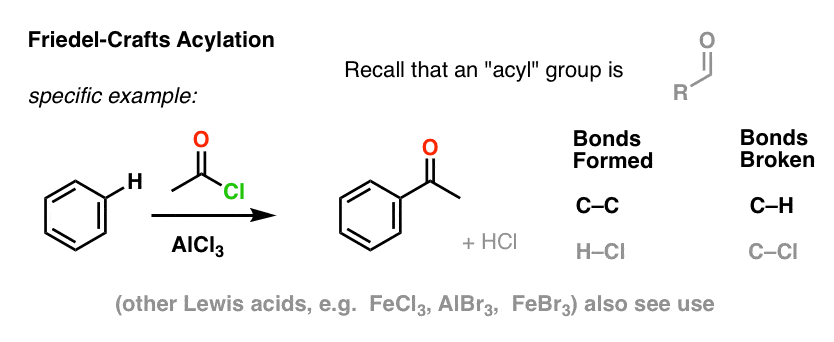
The Friedel-Crafts Acylation is similar, but we start with an acyl halide. Addition of our Lewis acid results in the formation of C–C and breakage of C–H.
8. Summary: The Six Key Electrophilic Aromatic Substitution Reactions
We’ve shown six key electrophilic aromatic substitution reactions (chlorination, bromination, nitration, sulfonylation, and the Friedel-Crafts alkylation and acylation) and that they all involve the breakage of C-H and the formation of C-E (where “E” is the electrophile in question).
But knowing the bonds that form and break is just the beginning.
Here’s some questions we’d like to know the answer to:
- How do substituents on benzene affect this reaction? For example, how might these reactions be affected if we performed them on methylbenzene? or phenol? or chlorobenzene?
- How do electron-donating or electron-withdrawing substituents affect the rate of the reaction?
- With benzene, only one mono-substituted product can possibly be formed. But what if we start with a mono-substituted product and do an electrophilic aromatic substitution on it? Where do the substituents end up?
- How does the reaction work? How do we explain the formation of C-E and the breakage of C-H ?
- What about other aromatic groups (beyond benzene). How do electrophilic aromatic substitution reactions on pyrrole, pyridine, naphthalene, or other groups compare?
In the next post, we’ll discuss the first question: the effect of electron-donating and electron-withdrawing substituents. We’ll talk about activating and deactivating groups.
Next Post: Activating and Deactivating Groups
Thanks for reading!
Many thanks to Matthew Knowe for assistance with this post.
Notes
Quiz Yourself!
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
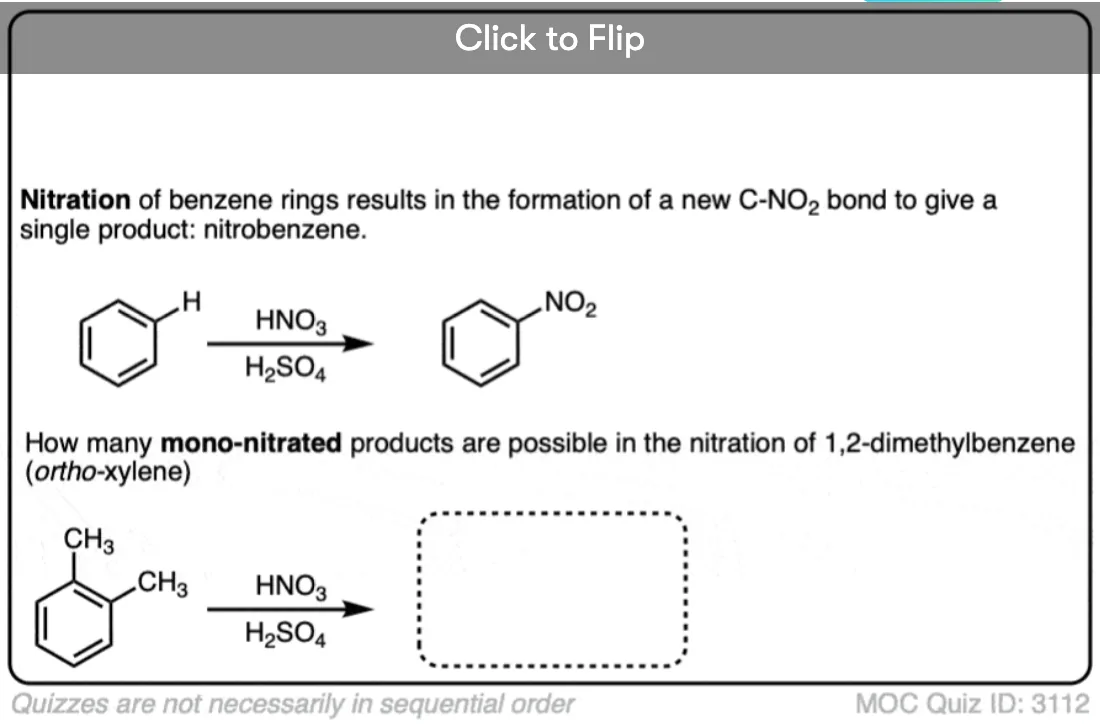
Become a MOC member to see the clickable quiz with answers on the back.
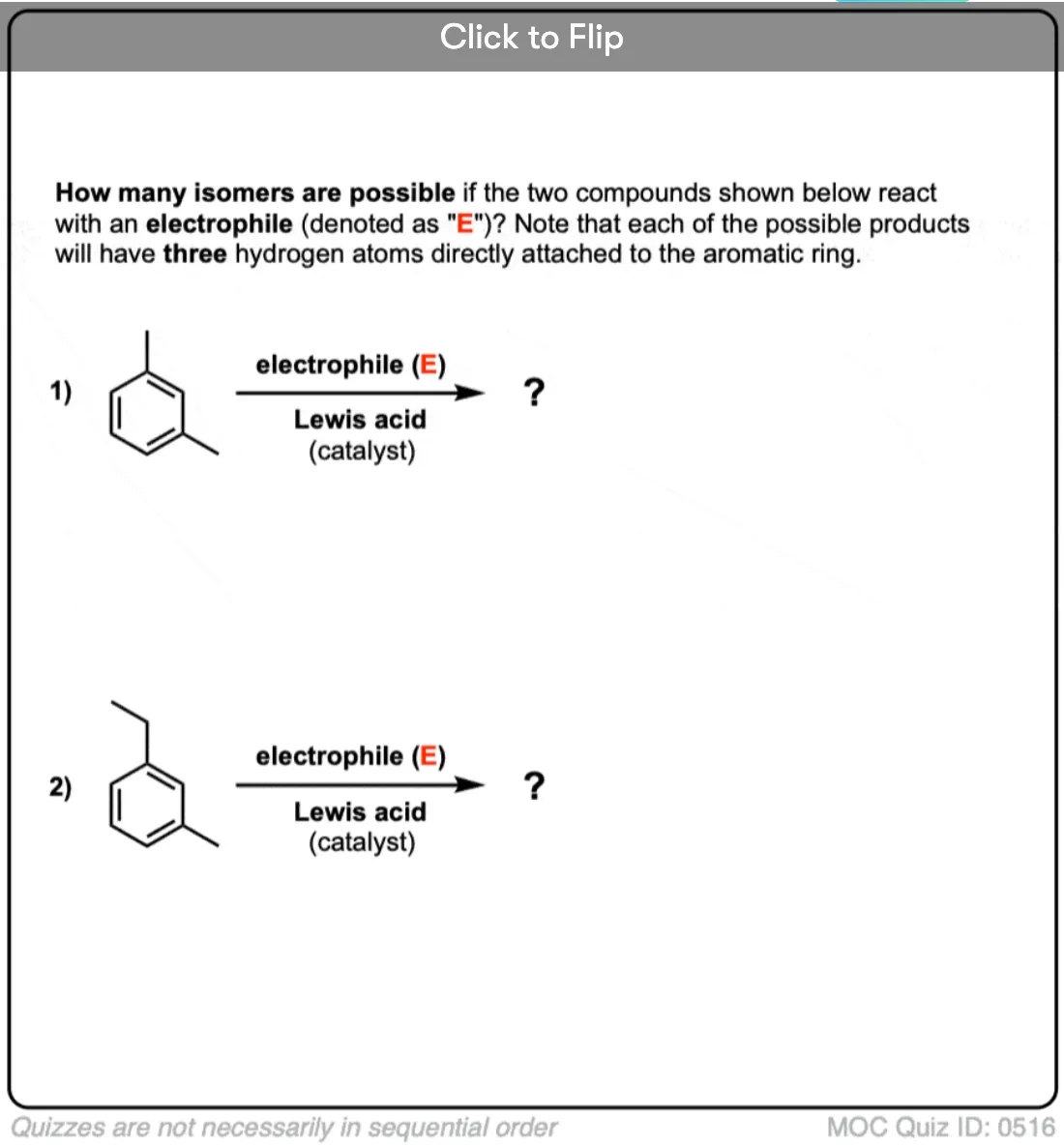
Become a MOC member to see the clickable quiz with answers on the back.
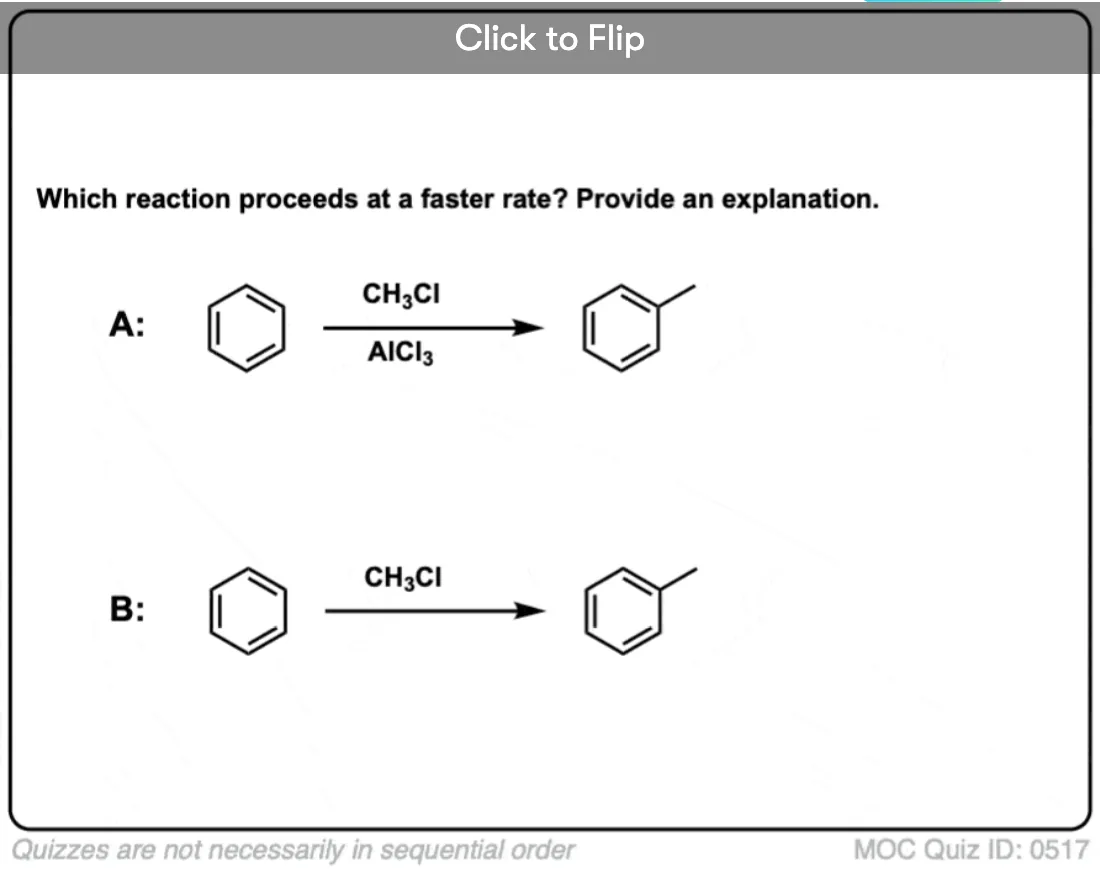
Become a MOC member to see the clickable quiz with answers on the back.
It’s because the molecule lacks a plane of symmetry.
I truly loved the way you explain!!! Thank you continue through…
Hello, I don’t understand how the quiz question on ”How many isomers are formed” lead to the c(ch3)3 substituted benzene having four isomers?
Please also include about addition reactions on multiple aromatic systems like anthracene, biphenyl etc. It will be very much helpful
I have a question. I’m currently working on the project. It consists of getting dicoumarol from coumarine from cinnamon. One step is adding OH group on fourth carbon atom so I can get 4-hydroxycoumarin. I have no idea and I would be so grateful if someone answered me their advices. Please, it’s really urgent. Thank you in advance
Please provide order of electrophilic reaction in different aromatic compounds (benzene and heterocyclic)
That sounds like a good topic for you to research!
Fine but not enough. Disubstituted compounds should be discussed
Covered in a later post!
Just one Suggestion
Plz organize the topics in an order so that the readers can go through them serial wise as it is very difficult for us(students) to search for the next topic and is time consuming as well.
If there’s a provision of that plz accept my apology as I am new to this website.
But the lessons are excellent..!!!! ? The explanation is Nice….
Thank you
Yes – in progress!
Amazing!
“Since Br2 by itself is not a strong enough electrophile to react with benzene at a reasonable rate…” – it is usually true, but the reaction is fast with concentration of Br2 being high (without any catalysts) [JCC 2016, 37, 210–225; DOI: 10.1002/jcc.23985].
Sure – it can happen (especially with electron rich substrates), but generally speaking, you want to use a catalyst.