Dienes and MO Theory
The Diels-Alder Reaction
Last updated: June 2nd, 2026 |
The Diels-Alder Reaction Is Awesome
Today we’ll introduce a reaction I consider to be the most useful and powerful reaction in all of organic chemistry: the Diels-Alder reaction.
- In the Diels-Alder reaction, a diene combines with a pi bond (often called a “dienophile”) to give a new six-membered ring
- Two C-C sigma bonds and a C-C (pi) bond are formed, and three C-C pi bonds are broken
- In order for the Diels-Alder reaction to occur, the diene must be conjugated and must be in the s-cis conformation
- The Diels-Alder reaction is faster when there is an electron-withdrawing group on the pi bond (“dienophile”) and electron donating groups on the diene.
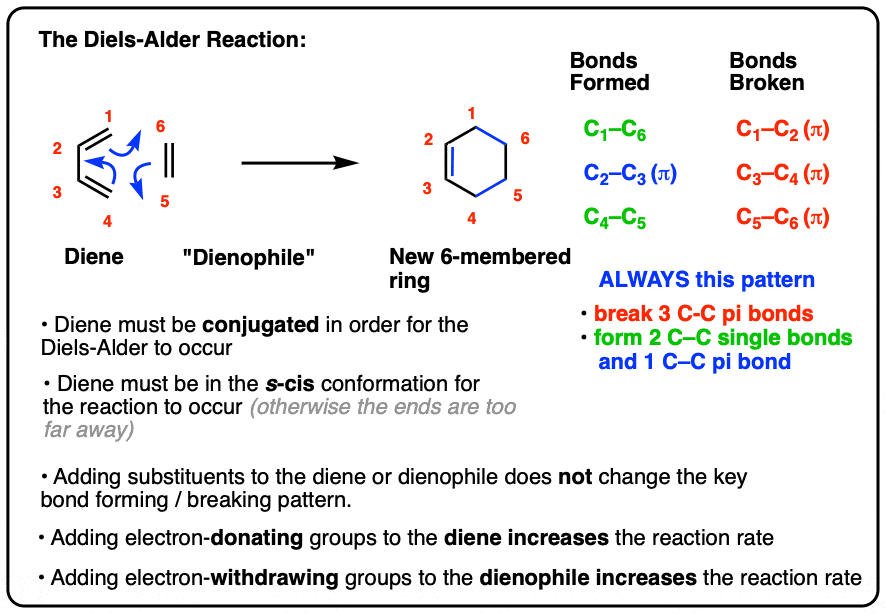
Table of Contents
- Chemists Unanimously Agree: The Diels-Alder Reaction Is Awesome
- The Diels-Alder Reaction
- Don’t Be Underwhelmed!
- The Basic Pattern of the Diels-Alder Reaction
- The Arrow-Pushing Mechanism of the Diels-Alder
- Four Key Things To Know About The Diels-Alder Reaction
- Four Key Things, Part 1: The Diene Must Be Conjugated
- Four Key Things, Part 2: The Diene Must Be In The s-cis Conformation
- Four Key Things, Part 3: Substituents On The Diene Or Dienophile Do NOT Affect The Bond-Forming / Bond-Breaking Pattern Of The Diels-Alder
- Four Key Things, Part 4. However, Substituents DO Affect The Rate Of The Diels-Alder. The Rate Is Increased By EWG’s On The Dienophile And Electron-Donors On The Diene
- Notes
- Quiz Yourself!
- Appendix 1:What About Other Ring Sizes?
- Appendix 2: Three Spectacular Examples of The Diels-Alder Reaction
- (Advanced) References and Further Reading
1. Chemists Unanimously Agree: The Diels-Alder Reaction Is Awesome
But don’t take some random blogger’s word for it. Here are three eminent chemists opinion on this reaction.

2. The Diels-Alder Reaction
OK. Are you ready to see it?
Prepare yourself.
Here it is:
My work is done here, folks.
Lesson over!
[drops mic]
3. Don’t Be Underwhelmed!
…wait. You’re not amazed? You’re not impressed? You are not blown away by the sheer power and beauty of this amazingly powerful process?
You are forgiven. Truth be told, I was a little underwhelmed myself when learned this reaction back in undergrad. [Nor was I as impressed as I should have been with the Cope rearrangement, which, when drawn in its simplest form, seemed pointless. In the name of Leo Paquette, I repent!].
That’s because instructors (myself included) tend to start with the most boring, simple, hand-holding examples possible so as to get the main pattern across, with the result that students miss out on seeing the really spectacular cases.
So, as much as I’d like to start with these complex and very beautiful examples of the Diels-Alder… we’ll start, as tradition dictates, in the shallow end of the pool.
[So for just a few ka-pow! examples of the Diels-Alder to spice up your day, see the footnote – Tetrodotoxin [Kishi, 1972]. Tetracyclines [Myers, 2005]. iso–Odoratin [Newhouse, 2017]
Let’s have a closer look at this extremely important reaction. First:
- The fundamental bonds formed/bonds broken pattern
- The simple arrow pushing mechanism
4. The Essential Pattern of the Diels-Alder Reaction
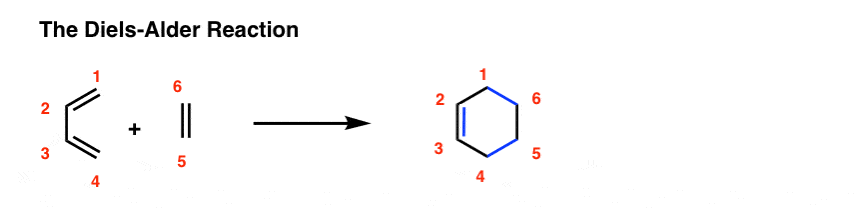
A Diels-Alder reaction brings together two components.
- One part we call the “diene“, which is comprised of two adjacent (i.e. conjugated) pi bonds.
- The second component is called the “dienophile“, which is to say “diene-loving”, and has at least one pi-bond.
What bonds form, and what bonds break here?
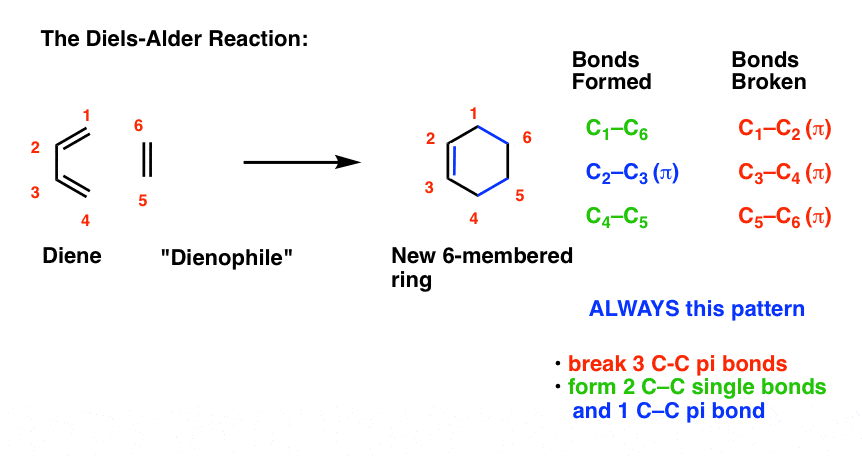
- Three pi bonds are broken. (C1-C2, C3-C4, and C5-C6) note – this is not IUPAC numbering, it’s just for us to keep track of each carbon for book-keeping purposes.
- Two new single bonds are formed (C1-C6, C4-C5)
- One new pi bond is formed (C2-C3).
Pay attention to this key pattern, because it repeats itself, over and over and over.
Indeed, every Diels-Alder that we learn about will follow this pattern.
Three pi bonds are broken; two sigma bonds and one pi bond are formed. Every. Single. Time.
The result is that a new six-membered ring is formed. (Every… Single…. Time! )
5. The Arrow-Pushing Mechanism of the Diels-Alder
My usual preference is to hold off on mechanisms until we’ve walked through some experimental facts, but here I will make an exception.
There are two ways to draw the flow of electrons (both correct): clockwise and counter-clockwise.
This flow of electrons is depicted with three arrows, which I’ve labelled A, B, and C. Pay attention what bonds form and break in each arrow.
![]()
You should be able to see that three C–C pi bonds are broken, and three new C–C bonds are formed (2 C-C sigma bonds and one C-C pi bond) no matter whether the electron flow proceeds in a clockwise or counter-clockwise direction.
[An aside: so far, almost every reaction you’ve seen in organic chemistry involves a nucleophile and an electrophile. The electrons always flow from the nucleophile to the electrophile. This begs a question. If the electron flow can be drawn as going in either direction, then what is the nucleophile and electrophile here? That turns out to be a very deep question – one which we’re not going to answer just yet! ]
6. Four Key Things To Know About The Diels-Alder Reaction
Having looked at the electron flow and the pattern of bonds that form and break, let’s continue with some very basic questions about the Diels-Alder.
Here are four key points:
- The Diene Must Be Conjugated
- The Diene Must Be In The s-cis conformation
- Substituents Do NOT Change the pattern of bonds formed/bonds broken…
- … but they do affect the rate!
7. Four Key Things, Part 1: The Diene Must Be Conjugated
The diene must be conjugated to participate in a Diels-Alder reaction. No conjugation, no Diels-Alder. So while 1,3-butadiene readily undergoes the Diels-Alder reaction, 1,4 pentadiene (below) does not.
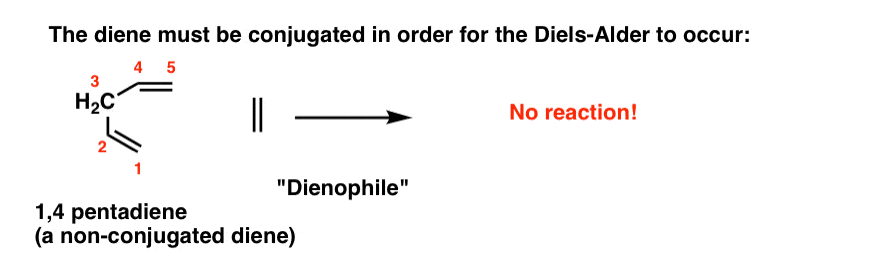
(If you’re not clear about what constitutes a “conjugated” diene, you might want to visit this post.)
8. Four Key Things, Part 2: The Diene Must Be In The s-cis Conformation
It’s not enough for the pi-bonds of the diene to be adjacent; the two C-C pi bonds must adopt a conformation where they are in the same plane (i.e. flat).
But even that isn’t sufficient. The diene must adopt a conformation where the two pi-bonds are oriented cis to the central C-C single bond in order for the Diels-Alder to occur. In other words, the diene must be in the s-cis conformation! [review: s-cis and s-trans]
For example, the Diels-Alder below occurs only when butadiene is in the s-cis conformation, and never in the s-trans.
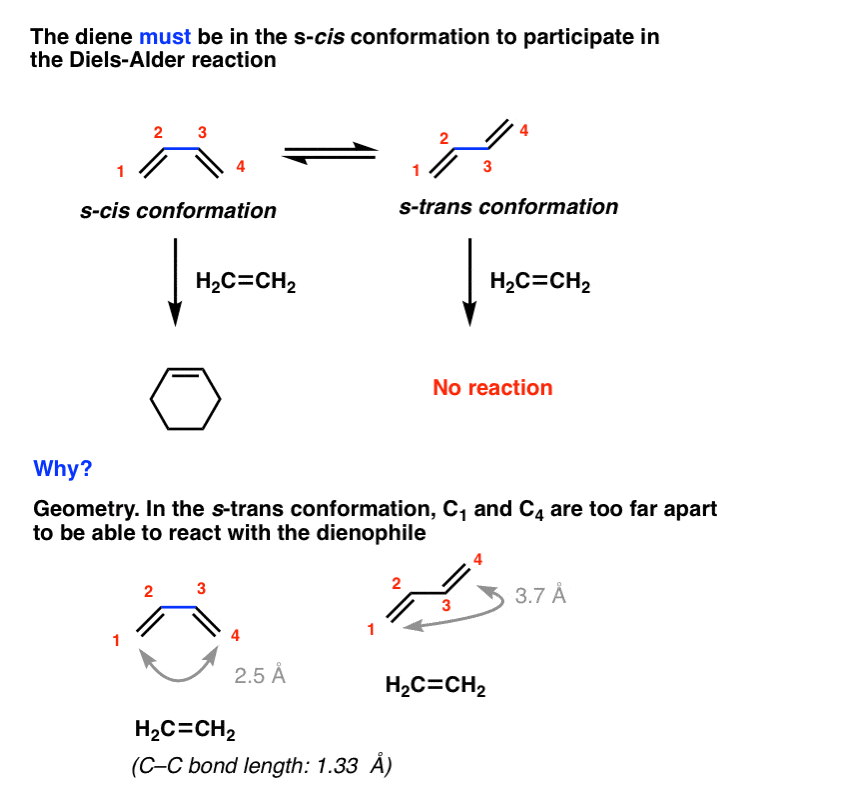
Why? In a word, geometry. In the s-trans conformation, the gap between the two ends of the diene (about 3.7 Å apart) cannot be bridged by the dienophile (about 1.3 Å long). This is only possible in the s-cis conformation.
There are many examples of dienes that are “locked” in the s-trans conformation, since rotation to the s-cis conformation would require the rupture of C-C bonds. These dienes can’t undergo the Diels-Alder.
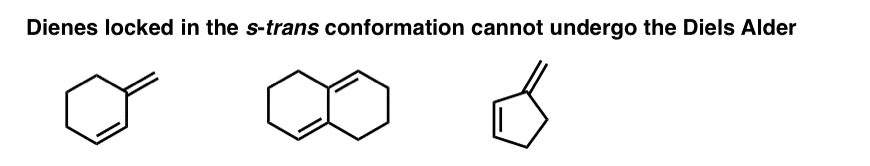
9. Four Key Things, Part 3: Substituents On The Diene Or Dienophile Do NOT Affect The Bond-Forming / Bond-Breaking Pattern Of The Diels-Alder
Adding substituents to either the diene or the dienophile does not affect the bond-forming/bond-breaking pattern of the Diels-Alder.
For instance, let’s tack a CH3 group (in red) onto C-1 of butadiene and sketch out the same reaction with ethylene.
This is what the product will look like:
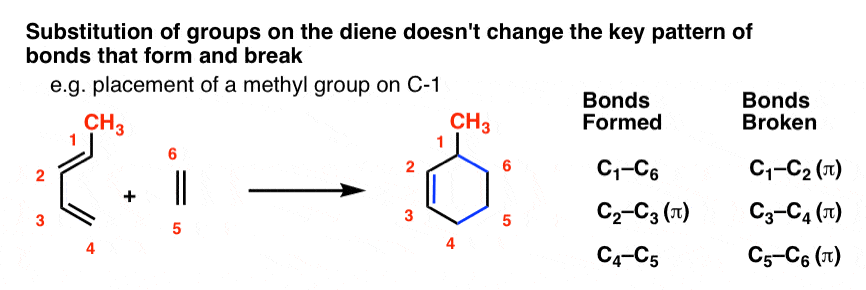
Note tha t the pattern of bonds that form and break is exactly the same as in the case of butadiene itself! The pattern doesn’t call for any of the bonds to the substituents to break, so we keep them all intact.
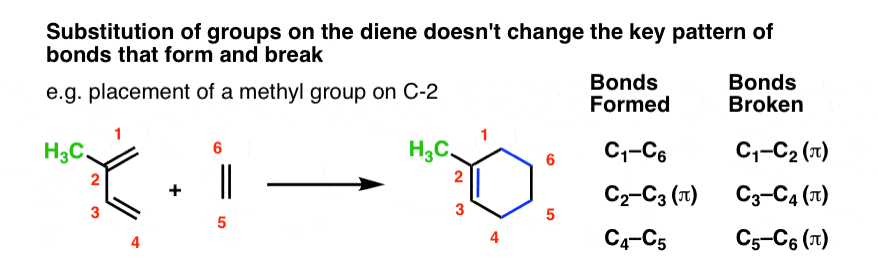
The same is true for substitution at C-2. The pattern of bonds that form and break is identical.
Although it might be hard to see at first, the same is true for much more complicated examples, several of which are shown in this appendix . All the extra cabbage on the diene doesn’t change the essential reaction pattern.
[The main complication of adding substituents, as we’ll see, is accounting for the stereochemistry of the product, a topic which we’ll cover in a subsequent post.]
Everything we just said about the diene also applies to the dienophile. Substituents on the dienophile don’t affect the key pattern of bonds forming and breaking. The substituents just come along for the ride.
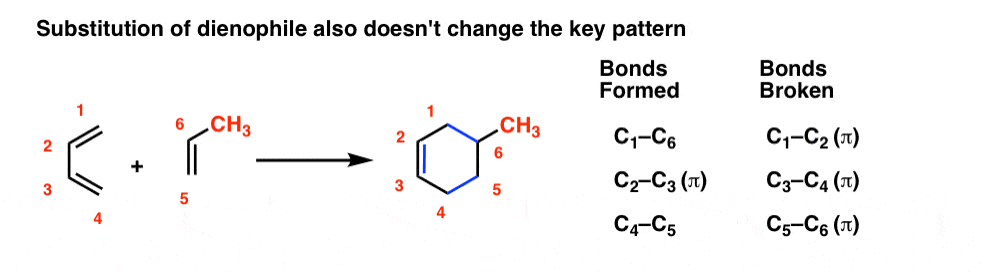
You might wonder: can we combine substitution on the diene and dienophile? You bet! It’s the same pattern as before.
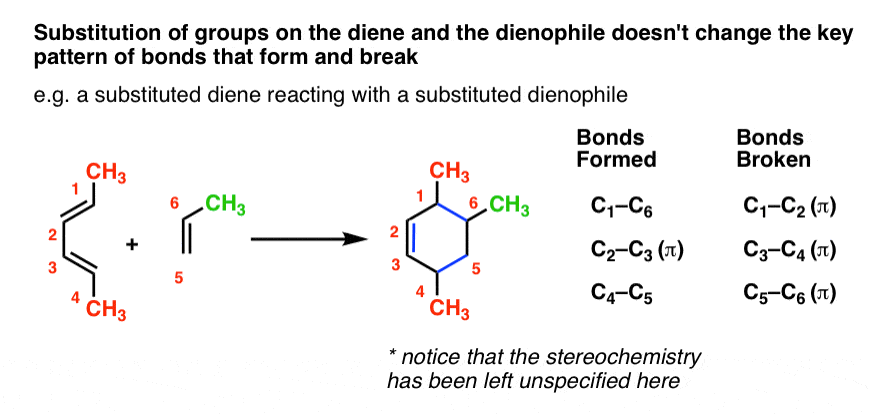
At the risk of repeating myself, let me say this again. The pattern of the Diels-Alder calls for breaking three pi bonds and forming two single bonds and a pi bond, resulting in a six-membered ring. Any bonds to the substituents on the diene/dienophile do NOT break. These substituents stay exactly the same.
10. Four Key Things, Part 4. Substituents DO Affect The Rate Of The Diels-Alder. The Rate Is Increased By EWG’s On The Dienophile And Electron-Donors On The Diene
While substituents may not change the essential pattern of the Diels-Alder reaction, they can greatly affect its rate.
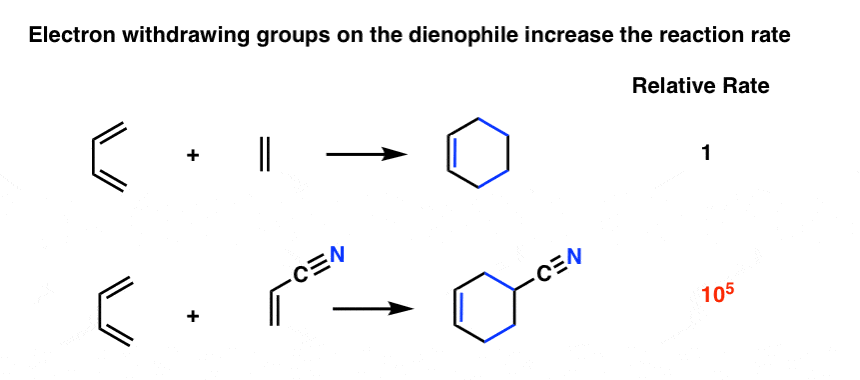
As it turns out the rate of the Diels-Alder is orders of magnitude faster if there is an electron-withdrawing group on the dienophile [Note 4] For example, replacing a hydrogen on ethene with the electron-withdrawing group CN (a nitrile) results in about a 105 increase in the reaction rate. [Note 5].
Other common electron withdrawing functional groups that will accelerate the Diels Alder reaction of dienophiles include aldehydes, ketones, and esters. In short, any functional group conjugated with the pi bond which can act as a pi acceptor will accelerate a Diels-Alder reaction with a typical diene.
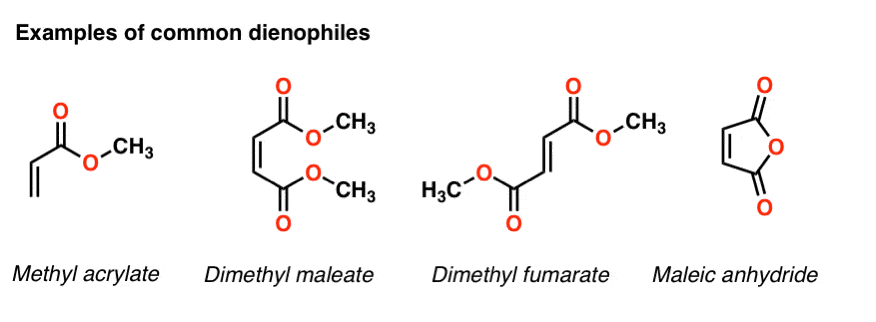
Making the diene more electron-rich also increases the rate. By “electron-rich”, I mean any substituent which can donate electron density to the diene. Alkyl groups (such as methyl and ethyl) are one example, as are pi donors such as oxygen and nitrogen.
For example, adding a methyl group on butadiene will increase the reaction rate by a factor of 3 to more than 100, depending on the dienophile [ ref] .
Next up: Cyclic Dienes and Dienophiles
Many students struggle with drawing out the products of Diels-Alder reactions with cyclic dienes and dienophiles. I was planning to address that topic in this post, but it’s getting too long. So we’ll cover that next time.
Thanks to Tom Struble for assistance with this post.
Notes
Note 1 For our purposes we’ll only discuss normal electron demand Diels Alder reactions, with an electron-rich diene and an electron-poor dienophile. However, there are examples of Diels-Alder reactions that proceed with electron-poor dienes and electron-rich dienophiles. These are known as inverse-electron demand Diels Alder reactions.
Note 2– These figures actually apply for cyclopentadiene, not butadiene, but the relative rates should be similar. For the experimental data see Carey and Sundberg A [page 641 on Google books] or direct from the literature, here. (German)
Note 3. Here is a 3-D visualization of a Diels-Alder transition state, courtesy of Rowan Labs. In this transition state, the length of the newly forming C-C single bonds are approximately 2.1-2.2 Å.
Quiz Yourself!
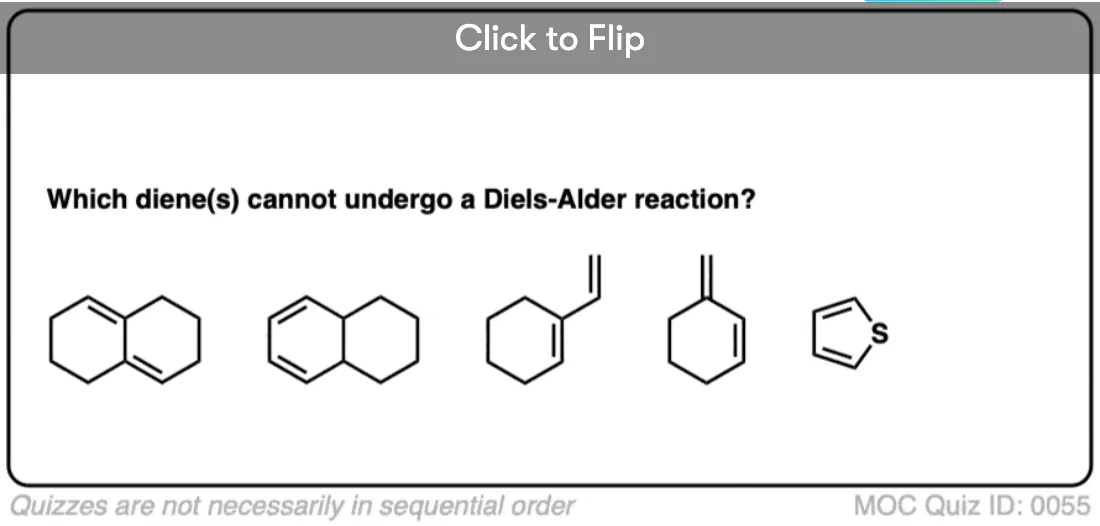
Become a MOC member to see the clickable quiz with answers on the back.
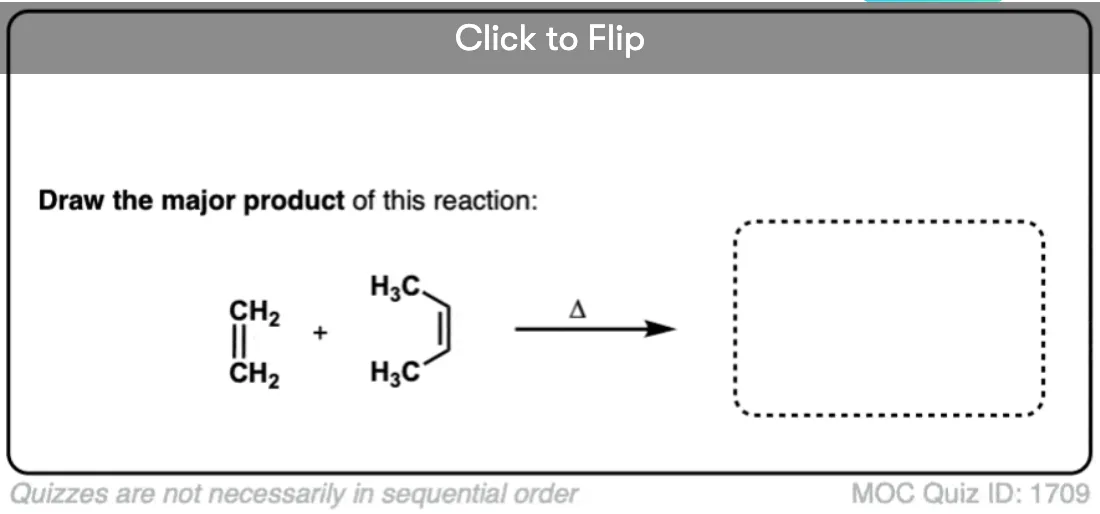
Become a MOC member to see the clickable quiz with answers on the back.
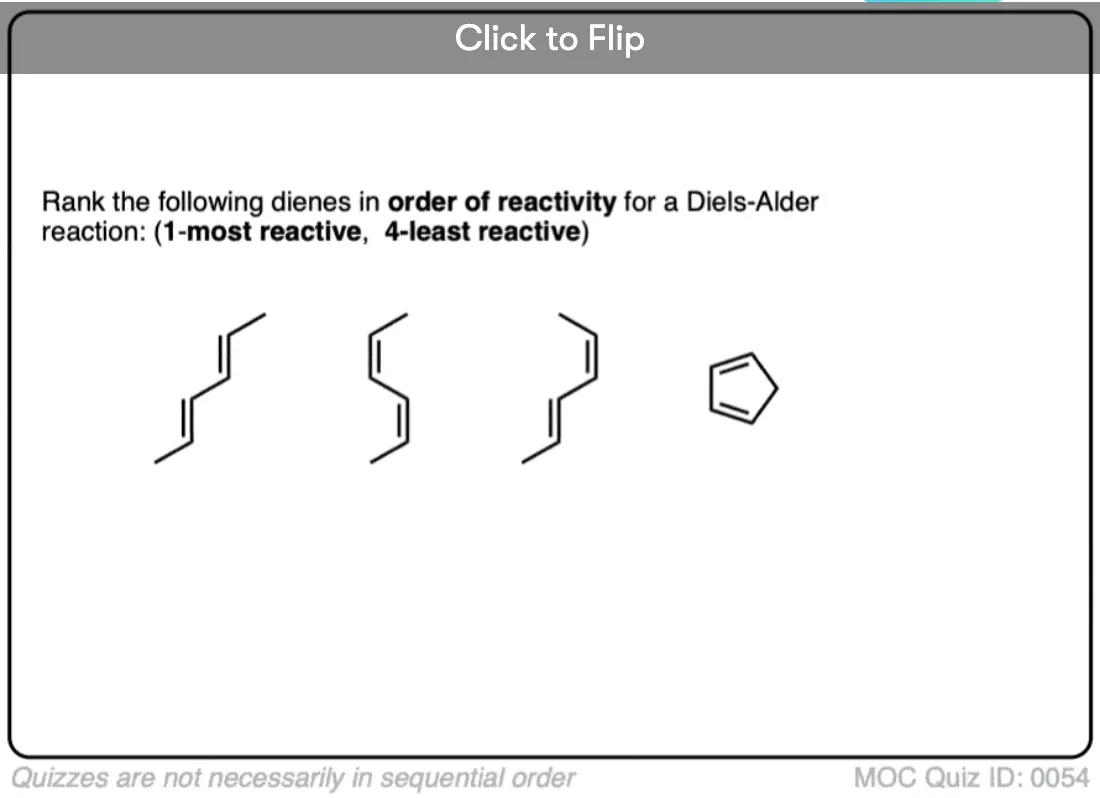
Become a MOC member to see the clickable quiz with answers on the back.
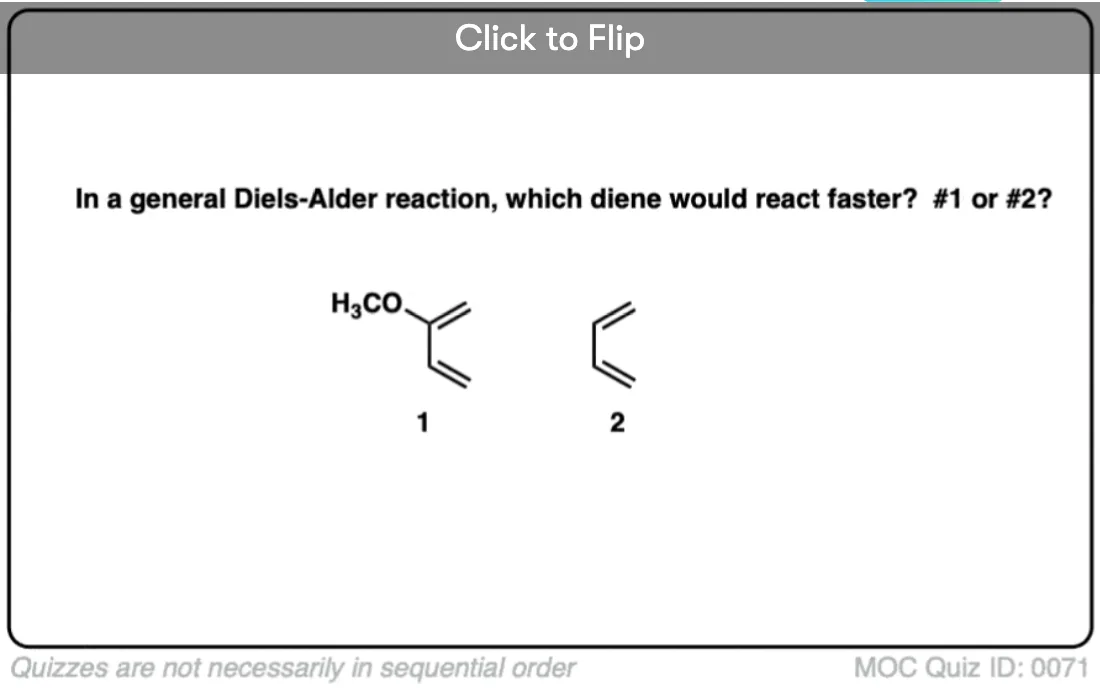
Become a MOC member to see the clickable quiz with answers on the back.
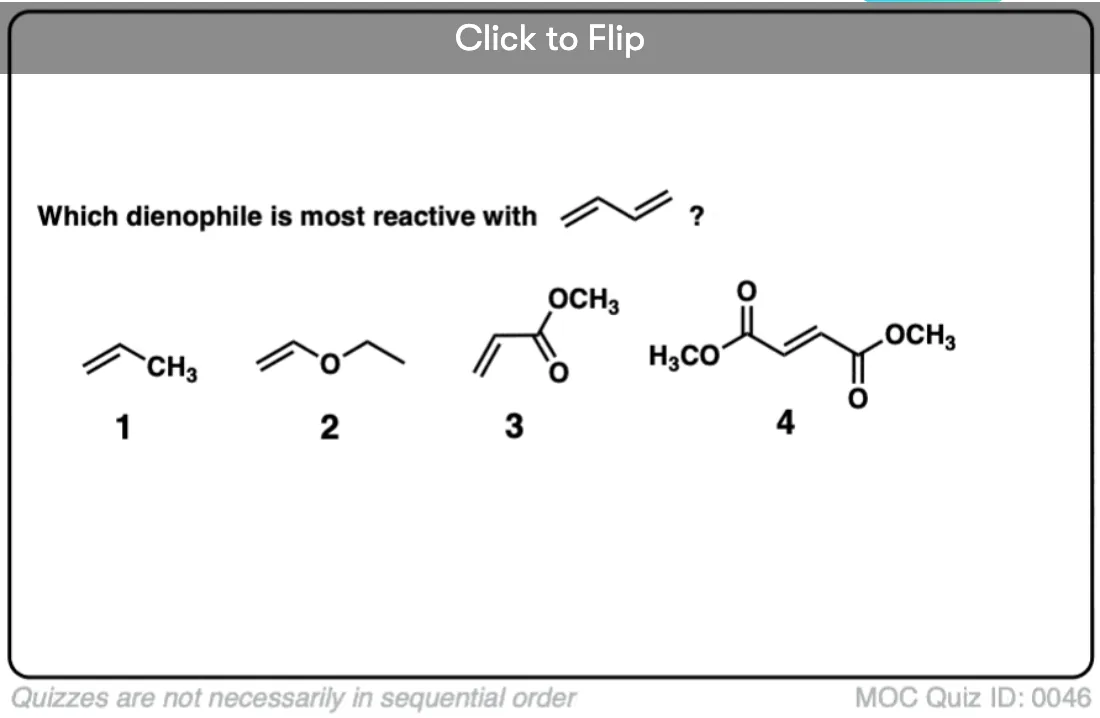
Become a MOC member to see the clickable quiz with answers on the back.

Become a MOC member to see the clickable quiz with answers on the back.
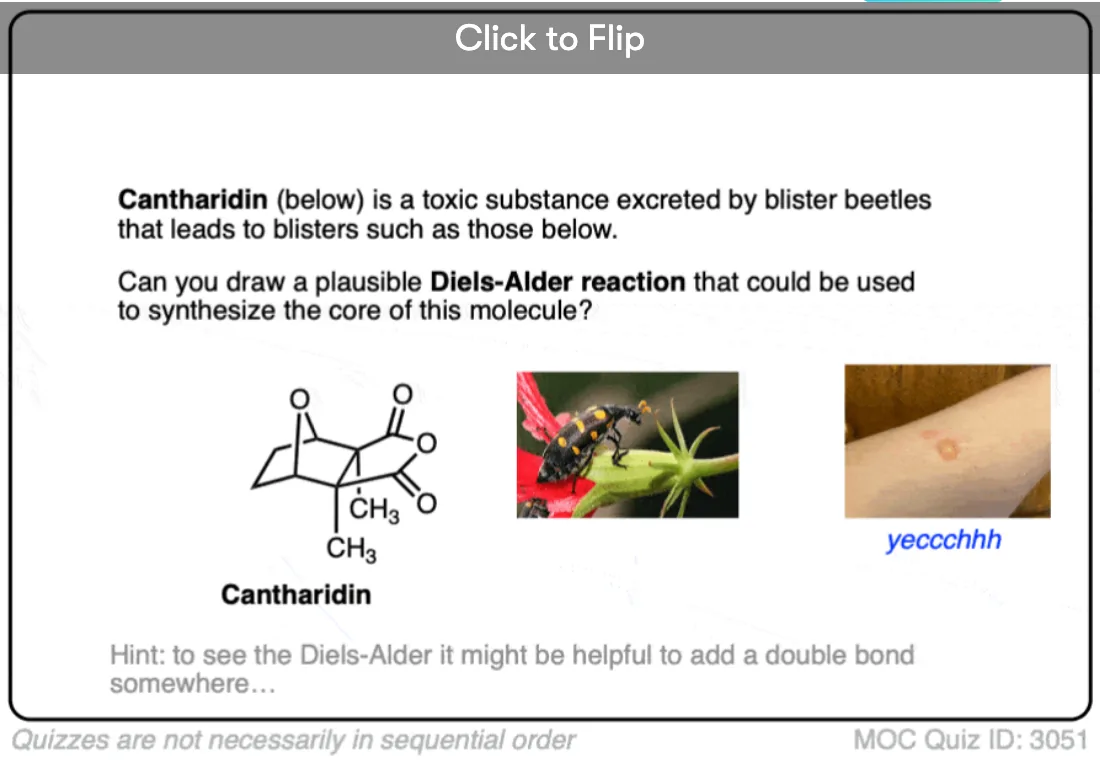
Become a MOC member to see the clickable quiz with answers on the back.
Appendix 1: What About Other Ring Sizes?
A Few “Dumb” Questions About The Diels-Alder That Are Actually Good Questions
1. Is This Type of Reaction Confined To A “Diene” and a “Dienophile”, or can it be used to make other sizes of rings too?
Great question. Up to this point we’ve been used to seeing highly charged nucleophiles and electrophiles.
The Diels-Alder is part of a family of reactions called “pericyclic reactions” where the two reaction components are relatively non-polar and do not generally go through charged intermediates such as carbocations
Specifically, the Diels-Alder reaction itself makes six-membered rings and only six-membered rings. A diene [4 pi-electron component] and a dienophile [2 pi-electron component] is required for the reaction to occur. For that reason the Diels-Alder is often called a [4+2] cycloaddition.
Are there other types of cycloadditions? Yes – although they aren’t nearly as common or important as the [4+2] cycloaddition.
For example, you might wonder: what happens if there’s no diene? Can two alkenes combine to make a four-membered ring?
Under the same conditions as the Diels-Alder (i.e. just with heat), no.
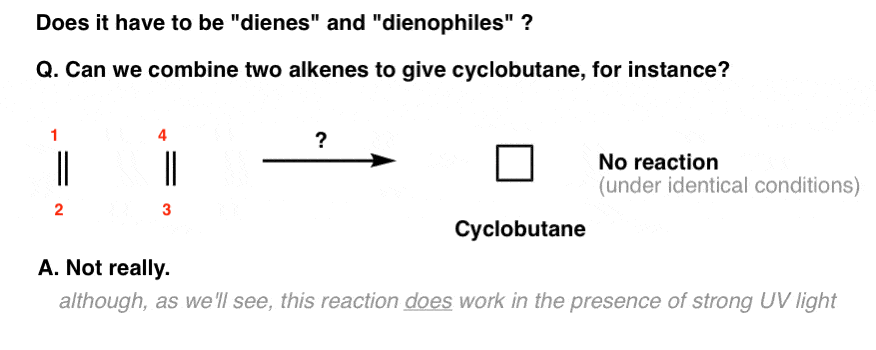
[We wrote a huge series of posts on the reactions of alkenes and you didn’t see this reaction. That’s for a good reason].
However, it is possible to get two alkenes to combine into a cyclobutane, if (and only if) strong ultraviolet (UV) light is applied.
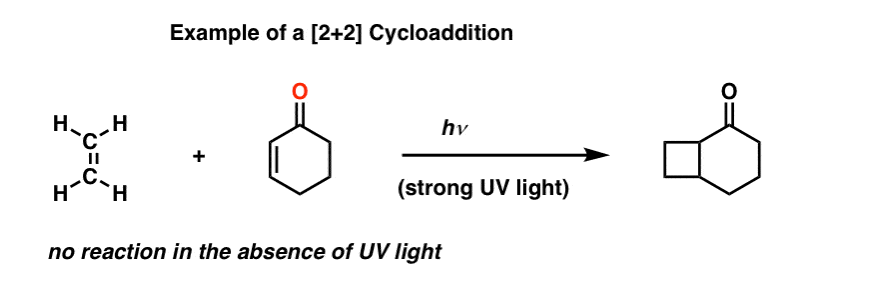
This is called a [2+2] cycloaddition.
Why is UV light required here but not for the Diels-Alder? Great question! It has to do with orbital symmetry, and we’ll get to that soon enough.
Along similar lines: what if there’s no dienophile? Can two dienes react?
In the absence of a dienophile, heating a diene under the same conditions does not give an eight membered ring (that would be a [4+4] cycloaddition, by the way).
What actually happens is that one diene combines with an alkene in a Diels-Alder reaction, forming a six-membered ring.
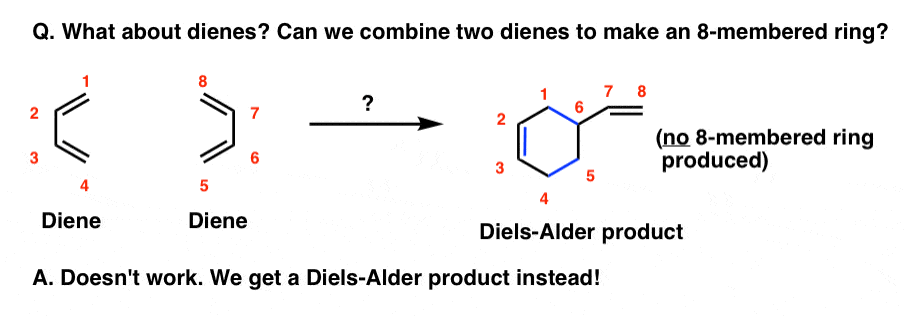
Other kinds of “higher-order” cycloadditions have been observed too, such as [6+4], [6+8], and even a [14+2] cycloaddition, but these are generally rare and quite contrived examples that pale in usefulness to the [4+2] case.
The point I’m trying to make here is that the combination of a diene + alkene giving a six-membered ring is particularly important and favoured.
2. Does The Dienophile Have To Be An Alkene?
The dienophile does not have to be an alkene specifically; the only requirement is that it contain a pi bond. It can also be an alkyne, for example. For example dimethylacetylene decarboxylate (DMAD) is an excellent dienophile, and after breakage of the C-C pi bond, an alkene remains. Even N=N pi bonds can participate in Diels-Alder reactions, so long as they are attached to electron poor groups, such as in diethylazodicarboxylate (DEAD). Yes, there is a reagent called DEAD.
Furthermore there are examples of C=O bonds participating in Diels-Alder reactions, although this gets into the territory of the inverse-electron demand Diels-Alder reaction, a topic we aren’t even close to discussing yet.
Appendix 2: Three Spectacular Examples Of The Diels-Alder Reaction
Advanced topic.
There are countless examples of the Diels-Alder reaction being used for the syntheses of natural products. I picked three below.
The examples will look complex to a beginner – and they are. However, the key point I want to hammer home is that the pattern of bonds that form and break in all of these reactions is NO DIFFERENT than the pattern we see with good ol’ butadiene and ethylene.
It takes about three years of intense training to obtain a licence to prepare pufferfish (Fugu) in Japan. Why is that? Because the liver and other selected organs of the pufferfish contain the deadly nerve toxin tetrodotoxin, and it takes tremendous practice to learn how to prepare fillets without contaminating the meat.
Tetrodotoxin has a wonderfully complex structure. The group of Yoshito Kishi at Harvard University first synthesized tetrodotoxin back in 1972 using a Diels-Alder to set the stereochemistry in the core 6-membered ring.
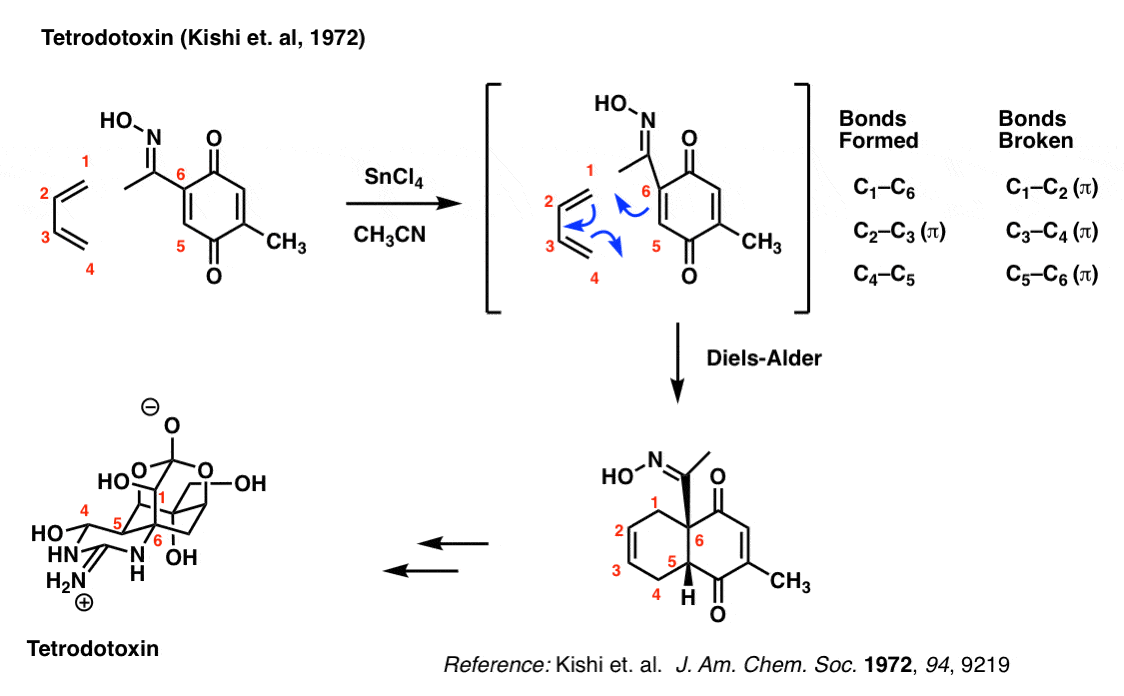
Try and look past the complexity to focus on the bonds that form and break here: you should be able to see that they are the exact same as those that form and break in the reaction of butadiene with ethylene.
The importance of the Diels-Alder here is that it set the stereochemistry of the C-5 and C-6 carbons (labelled in red), and formed a concave “cis-decalin” structure that allowed Kishi’s group to subsequently set the rest of the chiral centers in the molecule (7 total). This classic strategy, pioneered by Woodward, allowed for the synthesis of a dauntingly complex molecule at a time where there were very few methods for asymmetric synthesis.
You might wonder what the SnCl4 is for. That’s a Lewis acid that catalyzes the reaction between butadiene and the dienophile. Not only does Lewis acid catalysis make the reaction faster, but it also makes it more selective. Not a topic we’ll get into today.
Tetracycline (Myers and Charest, 2005)
Perhaps you’ve heard of the antibiotic tetracycline? The research group of Andrew G. Myers of Harvard University developed a platform for the synthesis of a diverse range of tetracycline antibiotics which relies on the Diels-Alder reaction.
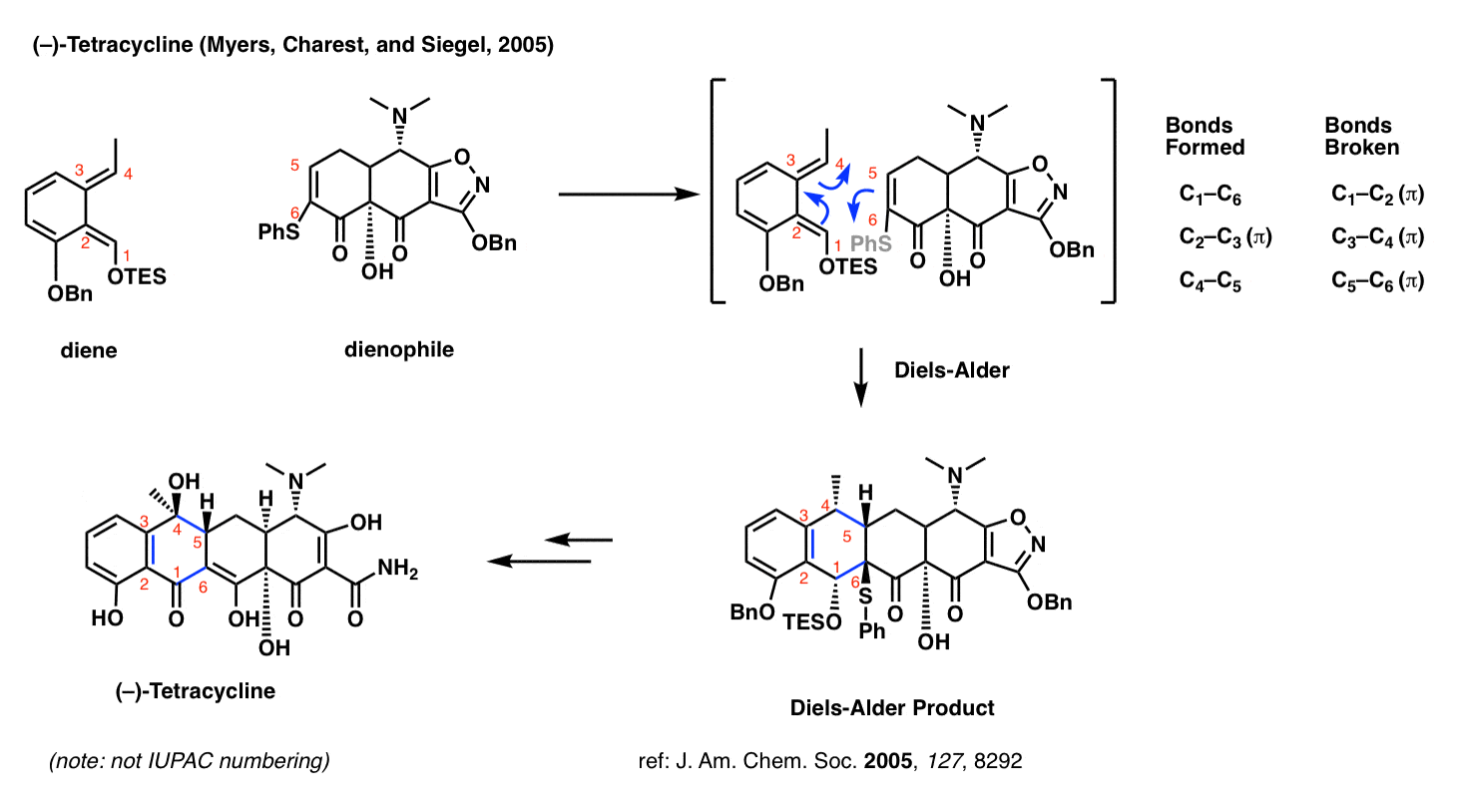
What makes the Diels-Alder so useful here? Two things. First, it sets the stereochemistry at C-5 (numbered in red – not IUPAC numbering). Secondly, and by far more important, is that the Diels-Alder allows one to join together two very complex fragments in a very predictable way.
Medicinal chemists commonly make a large number of different analogues of a promising drug candidate and test them under biological conditions to see which one performs the best. The beauty of the Diels-Alder strategy in the tetracycline synthesis is that it is convergent; one can make a complex diene and a complex dienophile and then bring them together at a late stage.
This strategy for tetracycline synthesis is the basis of a publicly traded company (Tetraphase) currently bringing treatments to the clinic.
iso-Odoratin (Newhouse and Schuppe, 2017)
The Diels-Alder isn’t some musty old reaction that is confined to the back pages of textbooks. It’s very much a part of current, cutting-edge chemical research. Just to demonstrate that this isn’t some reaction that’s buried in the past, here’s an example from January of this year, the synthesis of iso-odoratin (and other molecules) by the group of Tim Newhouse at Yale.
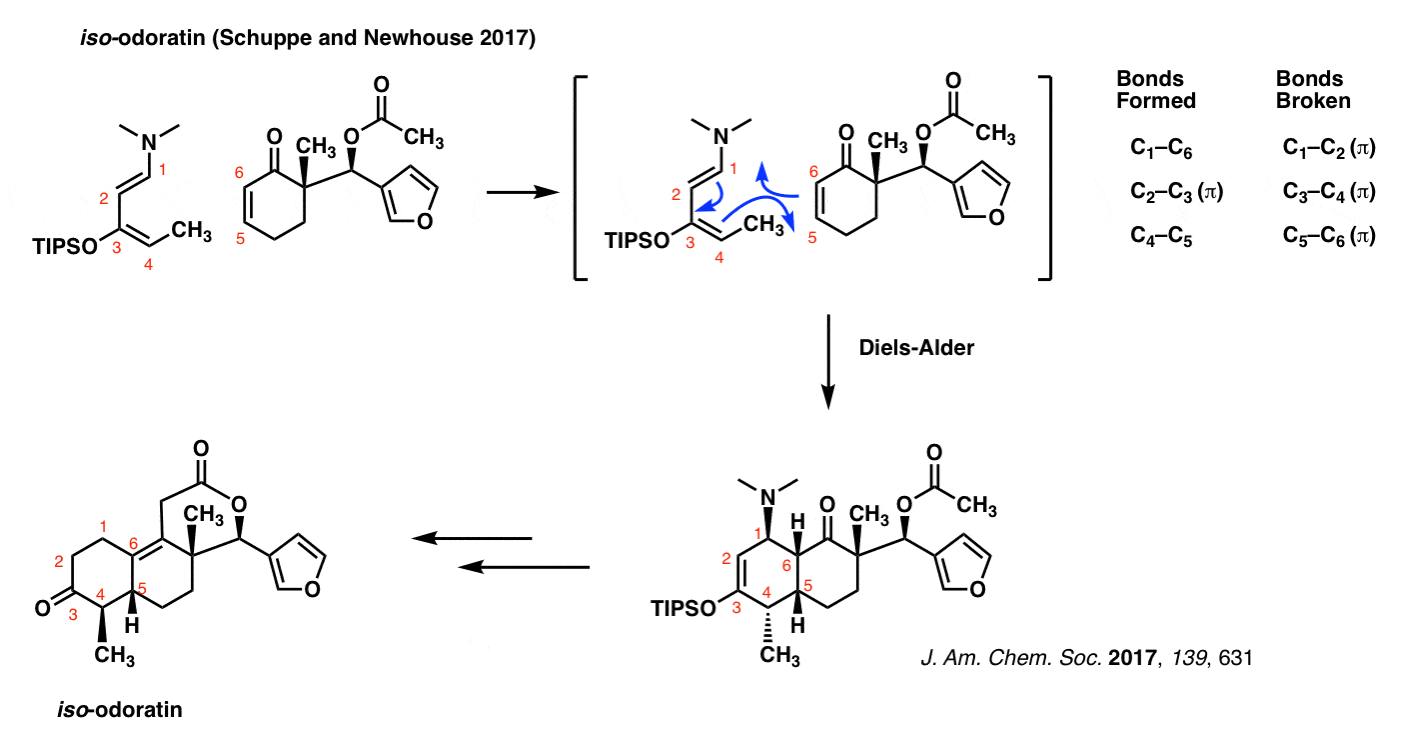
Note that as in all previous cases of the Diels-Alder, the pattern of bonds that form and break are identical.
The diene in this case is extremely electron-rich, possessing both an oxygen and a nitrogen (strong pi donors both!) directly bonded to the pi system.
Exactly why C-4 bonds with C-5 and not C-4 with C-6 (the regioselectivity, in other words) is a great question. We’ll address this in due course. [See: Regioselectivity in the Diels-Alder Reaction]
(Advanced) References and Further Reading
This one reaction has spawned a massive amount of research due to its versatility. The papers listed here are representative, but barely scratch the surface of the work that has been done in this area.
- Synthesen in der hydroaromatischen Reihe
Otto Diels and Kurt Alder
Lieb. Ann. Chem. 1928, 460 (1), 98-122
DOI: 10.1002/jlac.19284600106
This reaction is named after its discovers, Diels and Alder, who received the Nobel Prize in Chemistry in 1950 for this work. In this paper, they claim their territory in applying their reaction in total synthesis, stating “We explicitly reserve for ourselves the application of the reaction developed by us to the solution of such problems”. - The mechanism of the Diels-Alder reaction
R. B. Woodward, Thomas J. Katz
Tetrahedron 1959, 5 (1), 70-89
DOI: 10.1016/0040-4020(59)80072-7
Prof. R. B. Woodward was a legendary figure in organic chemistry, and this paper describes various theories as to the mechanism of the Diels-Alder reaction. - Diels-alder reactions with inverse electron demand. II. The reaction of benzamidine with π-deficient heteroaromatic compounds
P. Figeys, A. Mathy
Tet. Lett. 1981, 22 (15), 1393-1396
DOI: 10.1016/S0040-4039(01)90330-2
The normal Diels-Alder reaction proceeds best when the diene is electron-rich and the dienophile electron-poor. However, in certain cases, the opposite polarity is possible, and these reactions are known as inverse electron-demand Diels-Alder reactions. - The Diels–Alder Reaction in Total Synthesis
C. Nicolaou, Scott A. Snyder, Tamsyn Montagnon, Georgios Vassilikogiannakis
Angew. Chem. Int. Ed. 2002, 41 (10), 1668-1698
DOI: 10.1002/1521-3773(20020517)41:10<1668::AID-ANIE1668>3.0.CO;2-Z
Prof. K. C. Nicolaou (Rice) is a current leader in natural product total synthesis, and this review covers the applications of the Diels-Alder reaction in this area. - Hydrophobic acceleration of Diels-Alder reactions
Darryl C. Rideout and Ronald Breslow
Journal of the American Chemical Society 1980, 102 (26), 7816-7817
DOI: 10.1021/ja00546a048
This publication by Prof. Breslow (Columbia) is significant as it describes an unsual rate acceleration of the Diels-Alder reaction when carried out in water. This is ascribed to the hydrophobic effect, an entropic effect that forces the reactants closer together. - Useful diene for the Diels-Alder reaction
S. Danishefsky and T. Kitahara
Journal of the American Chemical Society 1974, 96 (25), 7807-7808
DOI: 10.1021/ja00832a031
This paper describes the synthesis and utility of a functionalized and reactive diene for Diels-Alder reactions. This diene is now commonly known as “Danishefsky’s diene” after its creator, Prof. S. Danishefsky (now at Columbia U.). [Worth noting that Danishefsky’s doctoral supervisor, Prof. Peter Yates, made an important advance in reporting Lewis acid catalysis of the Diels-Alder ] - New Strategies for Organic Catalysis: The First Highly Enantioselective Organocatalytic Diels−Alder Reaction
Kateri A. Ahrendt, Christopher J. Borths, and David W. C. MacMillan
Journal of the American Chemical Society 2000, 122 (17), 4243-4244
DOI: 10.1021/ja000092s
Enantioselective Diels-Alder reactions are also possible and are an active area of research. This paper by Prof. Dave MacMillan (now at Princeton) shows how one can use organocatalysis to achieve an enantioselective Diels-Alder reaction via a chiral iminium ion intermediate. - Conservation of orbital symmetry
Roald Hoffmann and Robert B. Woodward
Accounts of Chemical Research 1968, 1 (1), 17-22
DOI: 10.1021/ar50001a003
This is an extremely important paper. Conservation of orbital symmetry allows one to predict the stereochemistry of pericyclic reactions (such as the Diels-Alder reaction). This paper is a great introduction to the topic by its creators and introduces relevant terms (suprafacial, antarafacial, synrotatory, conrotatory, etc.). Prof. Hoffmann received the Nobel Prize in Chemistry in 1981 for this work, and would undoubtedly have shared it with Prof. Woodward that year (which would have been Woodward’s second Nobel) if he had not passed away untimely the previous year. - ENANTIOSELECTIVE, CATALYTIC DIELS-ALDER REACTION: (1S-endo)-3-(BICYCLO[2.2.1]HEPT-5-EN-2-YLCARBONYL)-2-OXAZOLIDINONE
S. Pikul and E. J. Corey
Org. Synth. 1993, 71, 30
DOI: 10.15227/orgsyn.071.0030
This procedure by Nobel Laureate Prof. E. J. Corey (Harvard) demonstrates an asymmetric Diels-Alder reaction by means of a chiral Al catalyst.
I think the link to the 3D visualization of the Diels-Alder transition site is broken. I’d love to see it. Thank you so much, you’ve helped me a lot!
Thank you James. What’s the German expression for “the moment when you realize you are much more lost than you thought you were?”
“Lakewood Avenue? That’s in Springfield. You’re in Dixon, son.”
We’re all to various degrees bumping into things in a dark room and gradually finding where the light switch is.
For this reaction, you list three pi bonds being broken — two on the diene, one on the dienophile.
But on the diene, since the two pi bonds (as drawn on a Lewis diagram) are conjugated, isn’t it actually just a single resonant conjugated pi bond, stretching from C1-C4 in the case of butadiene?
Saying it another way, aren’t we really breaking one bond on the diene, not two?
Also, in terms of rotation on the diene, I thought that atoms participating in a conjugated pi bond cannot rotate because the p-orbitals need to be aligned for the conjugation to occur — so the alignment of the p-orbitals is preventing the rotation between the C2 and C3 carbons.
So if the s-cis and s-trans dienes are conformers, and there is rotation around the C2-C3 bond, then that means there can be rotation around at least certain conjugated pi bond, right?
Thanks for the commment. It is not a single resonant pi bond. There is a small *barrier* to rotation about the C2-C3 bond (about 2-3 kcal/mol) but this is not the same as breaking a pi bond. For breakage of a pi bond you would need to have a barrier to rotation of about 60 kcal/mol.
Should we memorize this reaction? What is the reason for the reaction to undergo?
As a beginner, I am so fortunate able to eventually discover the DA reaction theory in detail. I can’t thank you enough.
Thank you so much for these excellent posts! They’ve helped me more than any single learning tool I’ve found so far- really nice content!!
Glad you’ve found it helpful Ian! Let me know if you find any typos/mistakes. Thanks
Why cyclooctadiene does not undergo dies alder
It is not immediately obvious. Relevant study: https://pubs.acs.org/doi/10.1021/acs.joc.5b00174
I just wanted to thank you. I’m in Orgo I. I read all assigned chapters religiously, attend all lectures, do all the practice questions in the textbook, but still struggle to understand what is being explained to me. I come to your site, and it’s as if you speak my language. I get it. I totally get it. I owe you, my passing grade. Thank you so much!
Alexandra, I am so glad to hear this. Thank you so much for sharing!
Is the 6+4 reaction between tropone and cyclopentadiene to give a nine membered ring not a Diels Alder reaction.. or are Cookson, Drake, Hudec and Morrison incorrect?
It’s not a Diels-Alder reaction. It’s a “higher order” cycloaddition. the “Diels Alder” refers specifically to the 4+2 case. If by Cookson you’re referring to the original report, I believe this was before Woodward and Hofmann generalized the selection rules. https://pubs.rsc.org/en/content/articlelanding/1966/c1/c19660000015#!divAbstract
Woodward Hoffman had predicted the 6+4 reaction prior to. When did it become a ‘higher order’ reaction. Mere words.
I stand corrected. They had indeed published the selection rules – 1965. https://pubs.acs.org/doi/10.1021/ja01087a034 . I believe Woodward assigned Ken Houk to look into 6+4 cycloadditions for his thesis project. Unfortunately the choice of heptatriene as the 6 pi component by Woodward/Houk was not nearly as fortuitous as the selection of tropone by your research group.
Thank you for all that you do. Helping pre med students everywhere
Glad you find it helpful, Coby!
Under the second section, you state that the diene must be in s-cis conformation otherwise a Diels-Alder reaction will not occur. Then, you show 1,3-butadiene in trans conformation and say no reaction happens. However, can’t 1,3-butadiene in trans rearrange to cis and undergo a reaction? The diene is not cyclic so it is not locked in trans postition.
With regards to the stereochemistry of the diene, you wrote that the diene must be in sigma-cis conformation in order react with the dienophile. What effect does ring size have on the reactivity of a cyclic diene locked in sigma-trans conformation?
For example, is the [4+2] cycloaddition between 3-methylenecyclohept-1-ene and a dienophile symmetry-forbidden or allowed?
Dear James,
Concerning the arrow-pushing mechanism, why is the arrow drawn from the dienophile to the diene? I was taught that arrows represent the flow of electrons. If the dienophile is electron-poor and the diene is electron-rich, shouldn’t the arrow be drawn the other way?
Nevermind, James. I’ve realized the Diels-Alder reaction is a one-step, concerted mechanism and as you mentioned, the electrons can move clockwise or counterclockwise.
I have another question.
If we react butadiene an ethene, we get the cyclohexene. Could the newly formed double bond act as a dienophile, which could react again with a diene? (and again to get a kind of polymerization)
I know that the dienophile should preferably have a EWG, but you stated an example where the ethene has a methylgroup attached, so it is not that far away.
Best regards,
David