Rearrangements
Carbocation Rearrangement Reactions (2): Alkyl Shifts
Last updated: May 29th, 2026 |
Alkyl Shifts In Carbocation Rearrangement Reactions, Including Ring Expansion
- Hydride shifts can sometimes occur when a more stable carbocation can be formed through migration of a C-H bond.
- If no hydride shift is possible that will result in a more stable carbocation, then it is possible that an alkyl shift can occur, where a C-C bond breaks and a C-C bond breaks. The net effect is the shifting over of the carbocation to the adjacent carbon.
- Alkyl shifts are most likely to occur when a quaternary carbon is adjacent to a secondary (or primary) carbocation
- Alkyl shifts adjacent to strained rings (e.g. cyclobutane) can result in ring expansion
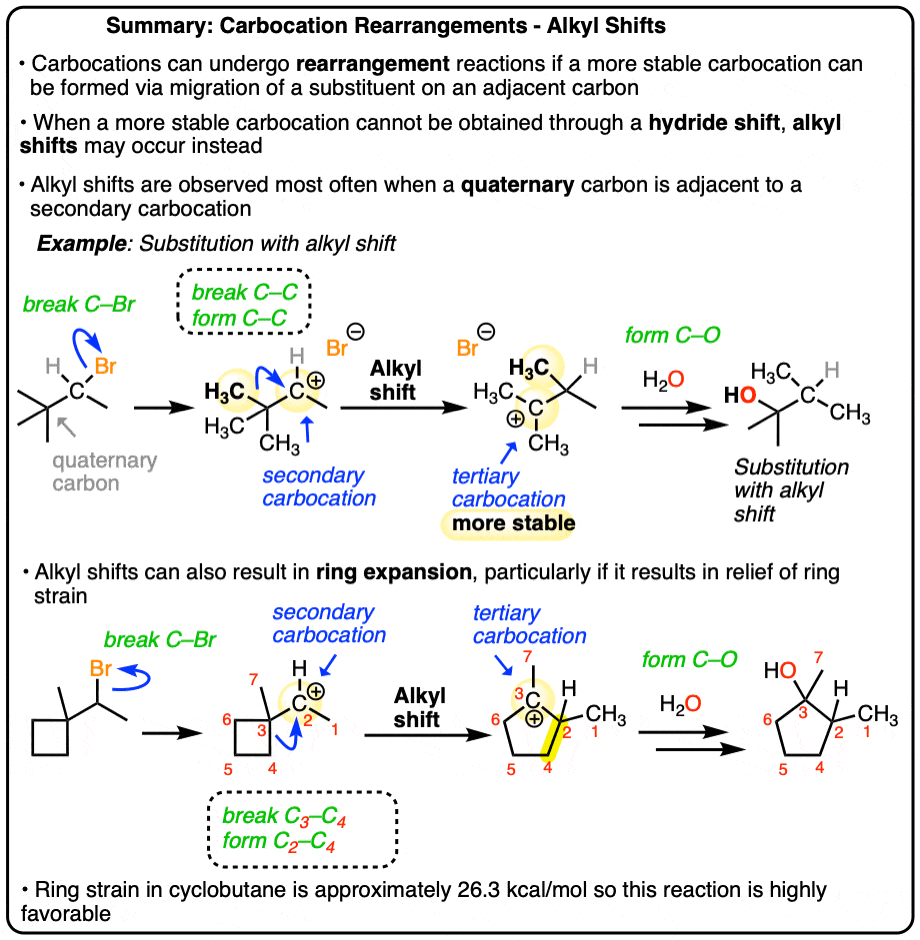
Table of Contents
- Alkyl Shifts Can Lead To More Stable Carbocations, Too
- The Mechanism Of Alkyl Shift Reactions
- Example of An SN1 With Alkyl Shift
- Ring-Expansion Reactions Also Involve Alkyl Shifts
- Notes
- Quiz Yourself!
- (Advanced) References and Further Reading
1. Alkyl Shifts Can Lead To More Stable Carbocations, Too
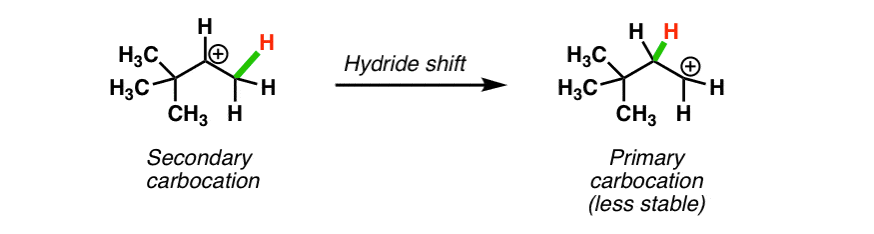
In the previous post we saw how certain carbocations can sometimes rearrange (through hydride shifts) to give more stable carbocations (See post: Hydride Shifts)
However, sometimes there are situations where a hydride shift would not lead to a more stable carbocation, but an alkyl shift would!
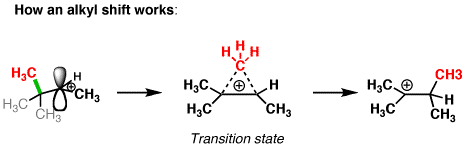
Take a look at this carbocation. If a hydride shift occurred here, we’d be going to a less stable (primary) carbocation! Not going to happen.
You might note something with this example, however: it is possible for a more stable tertiary carbocation to be formed.
How? If an alkyl group migrates instead! (Look at that red methyl group! )
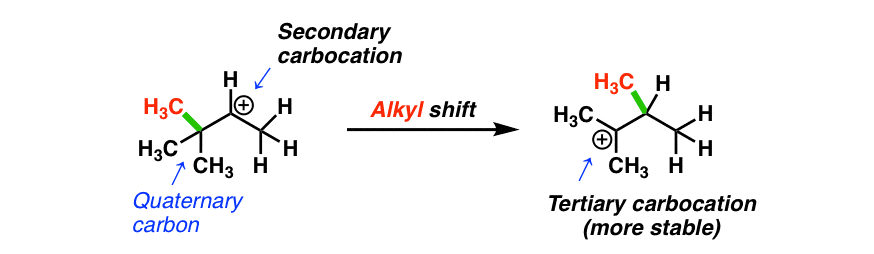
The most common situation where alkyl shifts can occur is when a quaternary carbon (that’s a carbon attached to 4 carbons) is adjacent to a secondary carbocation. (See post: Primary, Secondary, Tertiary, Quaternary)
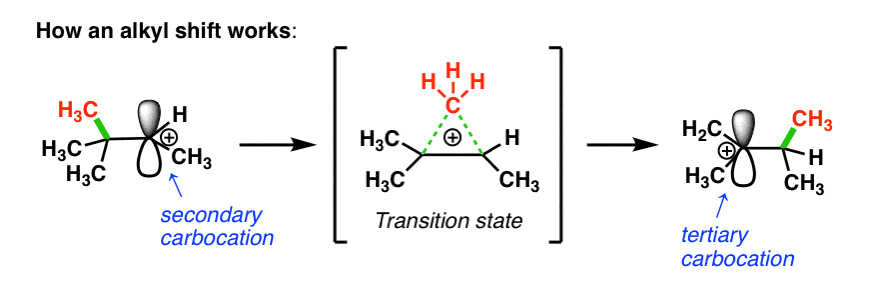
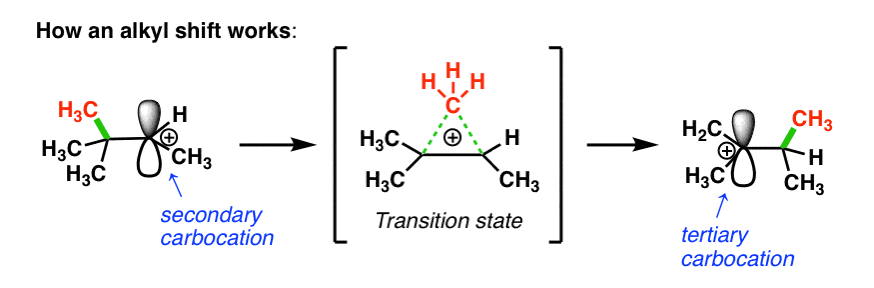
2. The Mechanism Of Alkyl Shift Reactions In Carbocation Rearrangements
How does this work? First, the pair of electrons from the C-C bond must align with the empty p orbital on the carbocation (side note: this means they have to be aligned in the same plane in order for orbital overlap to occur).
Then, as the pair of electrons from the C–C bond is donated into the empty p-orbital, one C–C bond begins to break and the new C–C bond begins to form.
In the transition state, there are partial bonds between the carbon being transferred and each of the two adjacent carbon atoms. Then, as one bond shortens and the other lengthens, we end up with a (more stable) tertiary carbocation. (See post: 3 Factors That Stabilize Carbocations)
{kind=link}
{kind=link}
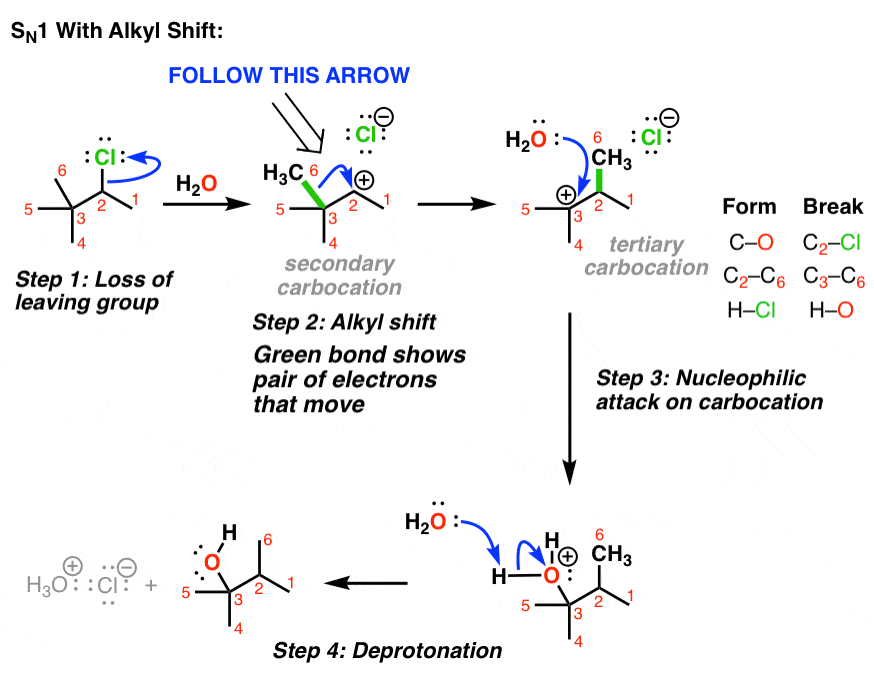
Rearrangements can potentially occur any time a carbocation is formed. That includes SN1 reactions (and as we’ll later see, elimination and addition reactions).
3. Example of An SN1 With Alkyl Shift
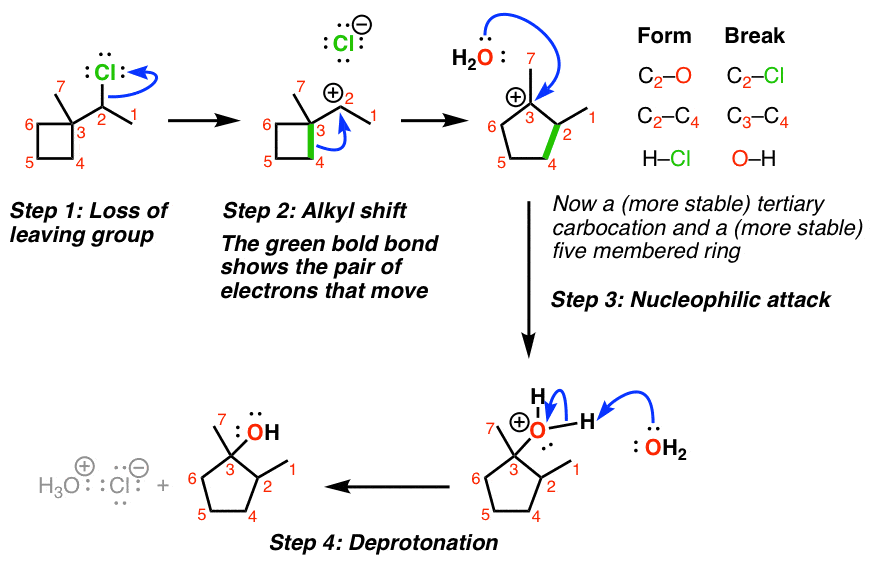
4. Ring-Expansion Reactions Also Involve Migration of Carbon
It doesn’t always have to be a methyl group that moves! One interesting example is when a carbocation is formed adjacent to a strained ring, such as a cyclobutane. (See: Ring Strain in Cyclopropane and Cyclobutane)
Even though the CH3 could potentially migrate in this case, it’s more favorable to shift one of the alkyl groups in the ring, which leads to ring expansion and the formation of a less strained, five-membered ring.
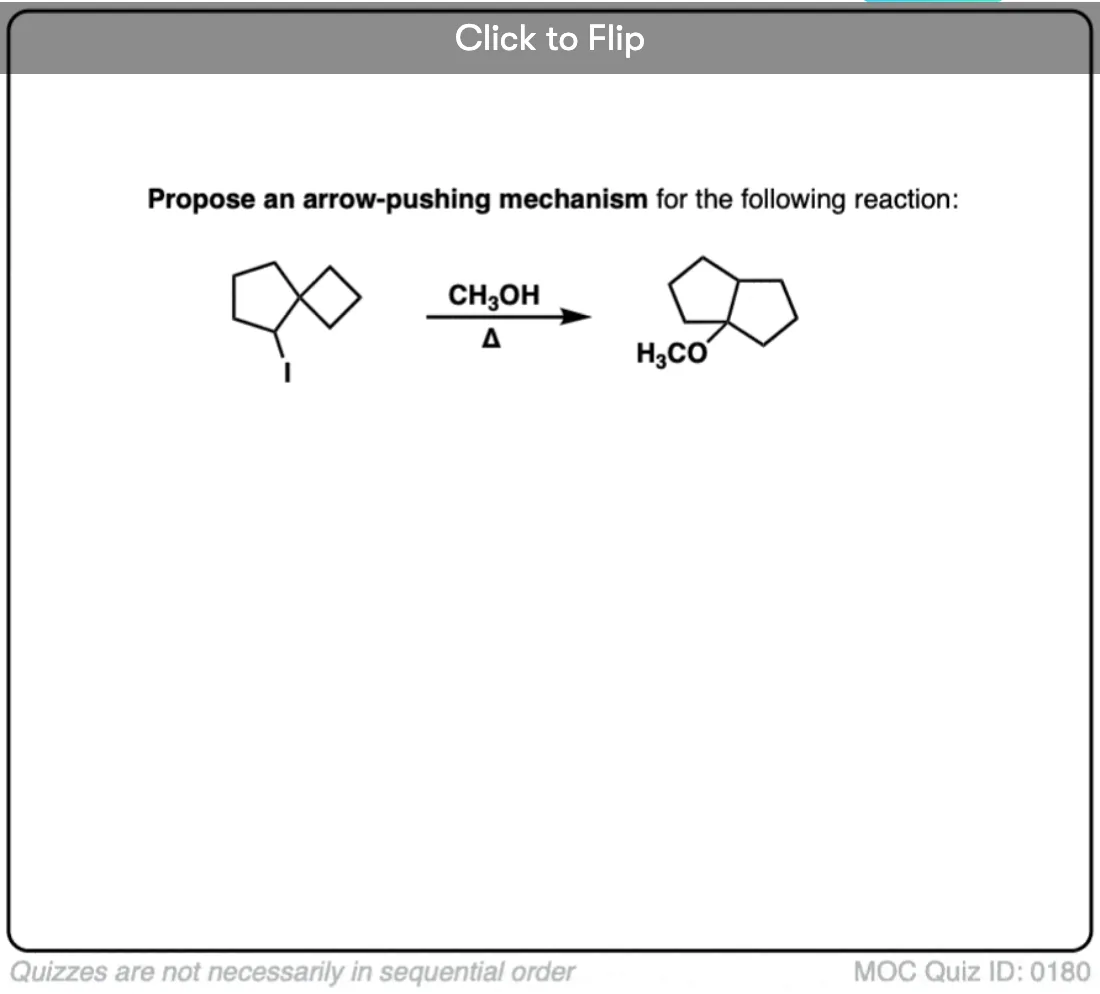
Here’s an example of an SN1 where migration of a carbon-carbon bond leads to ring expansion.
Notes
Quiz Yourself!
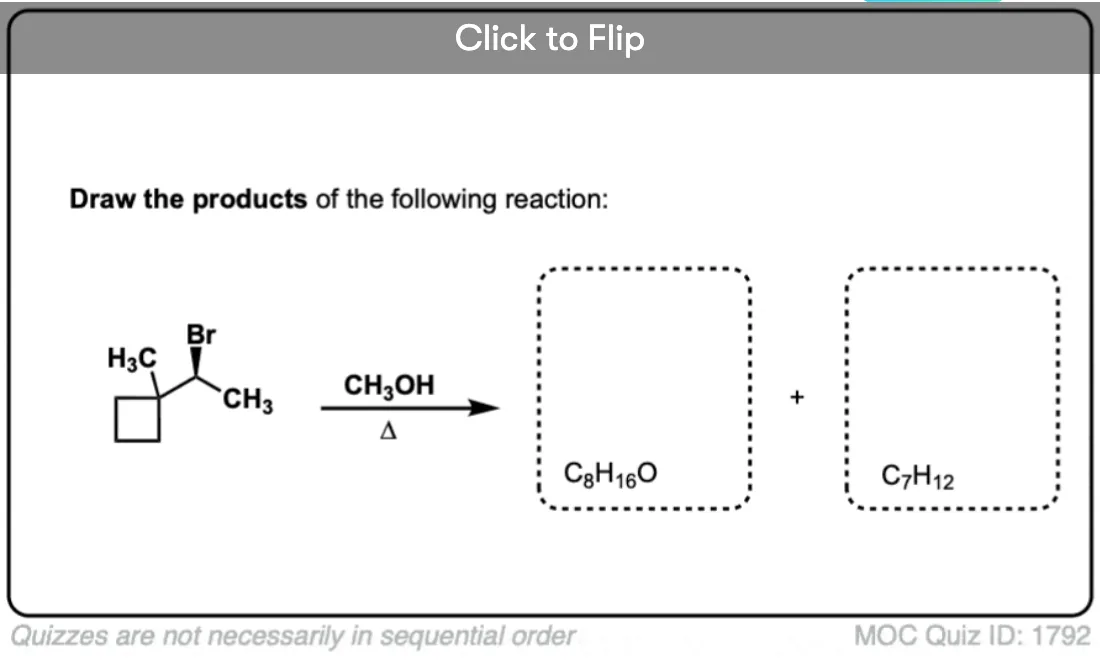
Become a MOC member to see the clickable quiz with answers on the back.

Become a MOC member to see the clickable quiz with answers on the back.
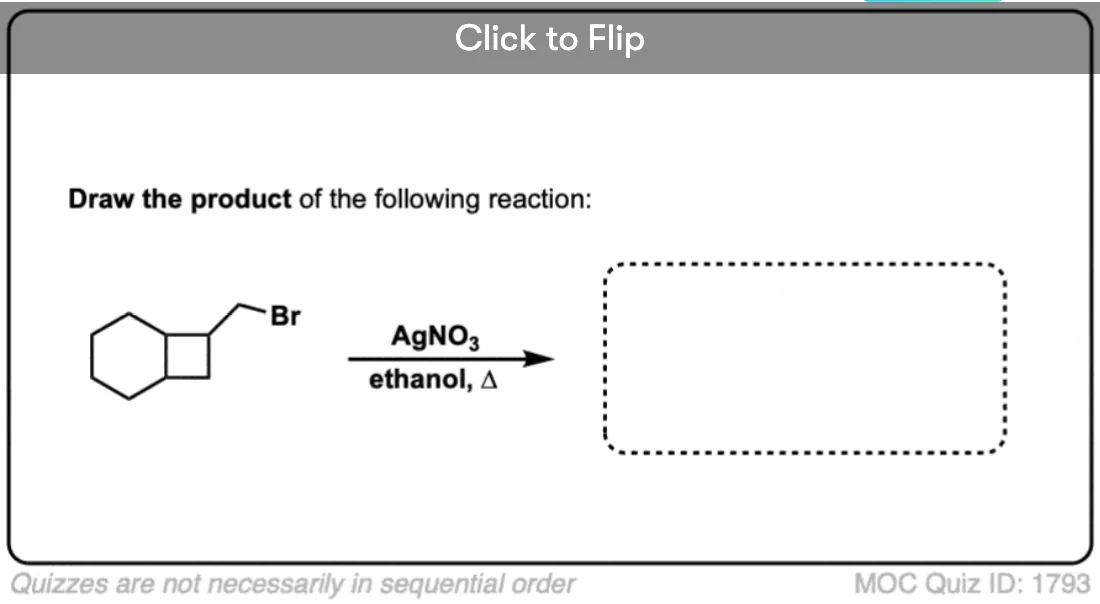
Become a MOC member to see the clickable quiz with answers on the back.
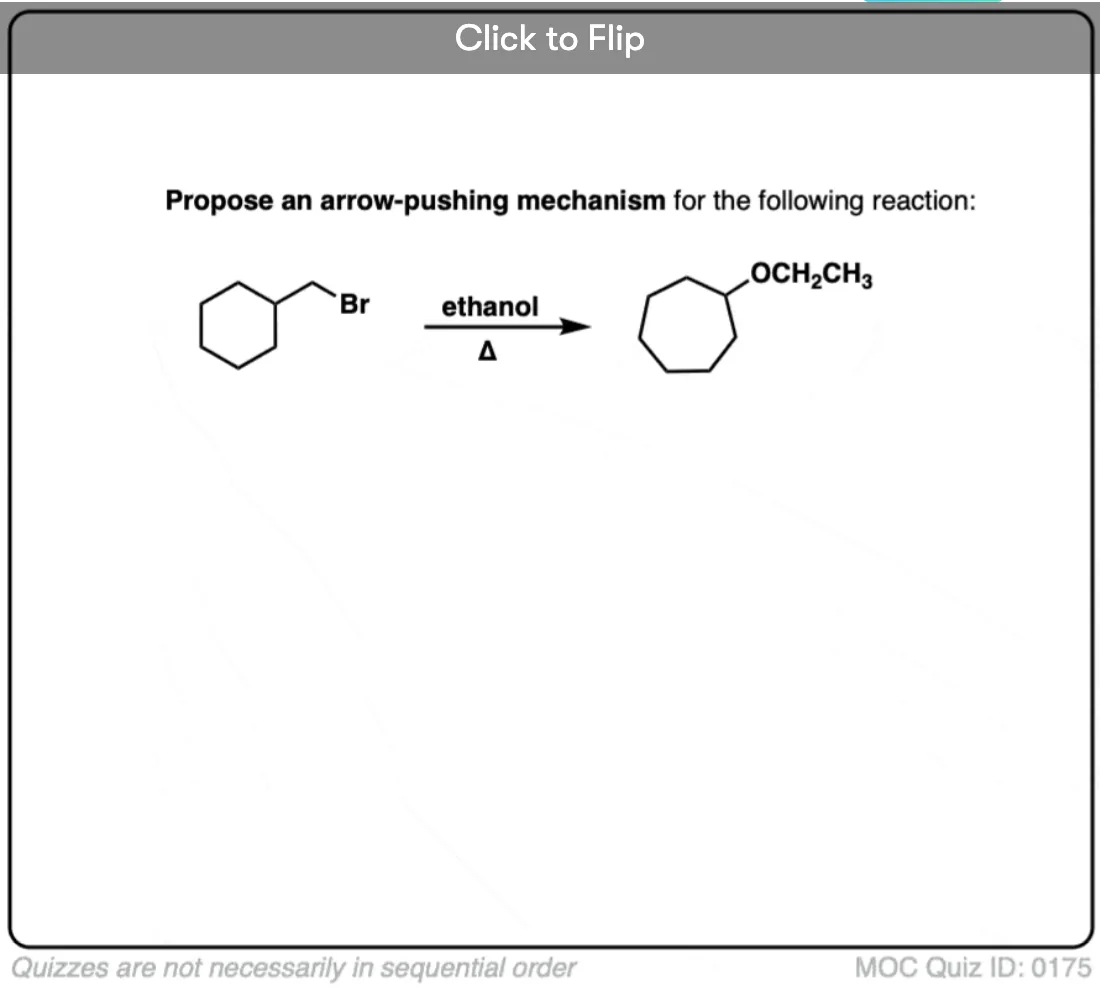
Become a MOC member to see the clickable quiz with answers on the back.
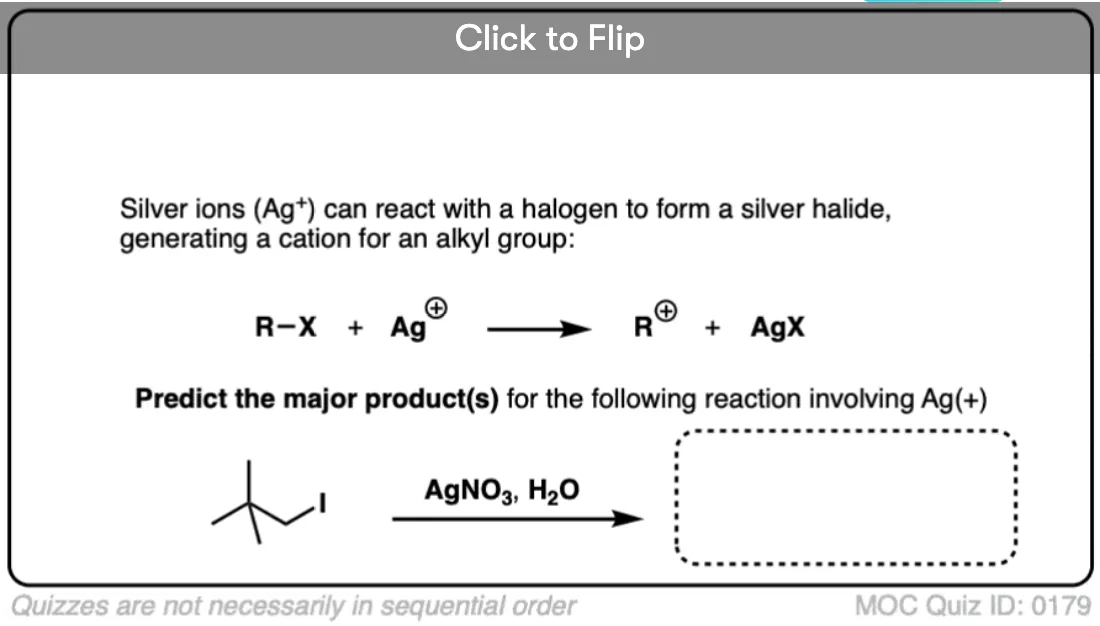
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading
[references]’,’Carbocation Rearrangement Reactions (2) – Alkyl Shifts
Angle strain…. i think
In an electrophilic addition reaction , say there is a carbocation which is on a 5 membered ring. It is in a position to expand to 6 membered, but on doing so its stability (via hyperconjugation) decreases. What product should we mark as major in that case?
There is really no driving force for ring opening. If it is going from a secondary carbocation to a tertiary carbocation in doing the ring expansion, then there would be a driving force, but the ring expansion going from 5- to 6- does not provide one.
Thanks for the post!
If in a situation both alkyl and hydride shifts can lead to more stable carbocations (example: secondary carbocation adjacent to tertiary and quaternary carbon) which one will give the major product(s) ?
Hydride shifts are faster.
Thanks for this wonderful website. I am not able to go for the membership plan, but all the information you’ve laid out for free is staggering and truly wonderful. I have learnt (and am learning) a lot from here.
I have a question too. If we take this molecule (https://www.wolframalpha.com/input?i=smiles+identifier+C1C%28%5BC%2B%5D%28C%29%28C%29%29C1), will it do ring expansion? Or will sigma-resonance stabilize it enough?
If there are possibilities of both tertiary carbonation and ring expansion then which one gets more preference?…which one forms?
In real life, both can occur.
In an exam-type situation, if you are faced with expanding a 4-membered ring to a 5-membered ring *or* doing a methyl shift to give a tertiary carbocation, you should expand out the 4-membered ring due to relief of ring strain.
There is very little relief of ring strain on going from 5- to 6-, and for that reason the two possibilities are a lot less clear cut in that case.
hi James if there were possibilities to form a tertiary carbocation in two ways in one way it’s an alkyl shift and another is a hydride shift, both leading to a stable carbocation (tertiary) which is likelyto occur …?
Almost certainly the hydride would migrate preferentially. It’s just easier from a principle of least motion standpoint.
Can ring expansion occur the other way around? I mean in a ring of Methyl cyclopentane. If there is a plus charge on the adjacent position of methyl. Will ring expansion still take place?
Do you mean ring contraction? Unlikely to go from five to four since four membered rings have a lot of strain.
If there is competing alkyl vs. hydride shifts that give equivalently stable carbocation, hydride shift will generally be favored since less molecular motion is involved.
Would you describe a ring expansion as exothermic or endothermic?
Generally, if one is going from a more strained ring to a less strained ring, the reaction should be exothermic since ring strain is being released.
Who is James why he is answering one?
Hi James, love your website and its worth every penny. We just had an interesting discussion in our study group regarding rearrangements and need an expert to weigh in. The BIG question: do rearrangements occur sequentially after carbocation formation or does it happen so fast that it appears as just one step? As an example: Hypothetically, imagine a substitution reaction at a primary carbon connected to a secondary carbon which is then connected to tertiary carbon. If you could form the primary carbocation, would the secondary then form, followed by the tertiary in a series of steps? In our discussion we imagined a reaction energy diagram with three humps, one for the activation energy of each carbocation intermediate and then a hodgepodge of products. We also considered that if this was the case a secondary carbocation would never result from a primary since they “never form”. On the other hand we also considered that the rearrangements happen simultaneously and so rapidly that they are coincident with the formation of “the most stable carbocation possible”. Finally, our Buddhist teammate said that we should not think in terms of duality so we broke it off and grabbed some beers and decided to ask you. (By the way we noted your example of lanosterol synthase. If it does happen sequentially that must be a real mess of side products)
Hi Len – it’s reasonable to postulate the existence of a transient secondary carbocation, but likely not a primary one. If rearrangement is happening to a primary site, likely that 1,2- shift is happening at the same time that the leaving group is being displaced so that at no point is there a “free” primary carbocation.
for a vinylcyclopentane reacting with H3O+, would it be better to do a hydride shift, or expand the ring structure?
HI Raeanne – I would start with the hydride shift, since the 5-membered ring is not very strained relative to (say) a 4-membered ring.
If we have choice,then which alkyl group we will shift,suppose we have methyl,ethyl and propyl,among this option wjow we will shift.
Hello sir. If we have cyclohexyl methyl bromide and we react it to water, will it go as per Sn1 reaction and cause a rearrangement( a hydride shift) or will it proceed directly via Sn2 mechanism?? In general, if a compound is 1° but it can go rearrangement, will it proceed via Sn1 or Sn2 mechanism??
The short answer is that if it’s primary, then you’re generally looking at an SN2 process since primary carbocations are unstable and difficult to form.
Sir,
If suppose during ring expansion, in the figure you have uploaded we have an oxygen atom at 4th position.Then which of the following bonds C3-C6 or C3-O4 will be broken for ring expansion?
What you are describing is the ring expansion of an oxetane into a tetrahydrofuran derivative. I would expect the C-C bond should migrate with simultaneous formation of an intermediate C=O pi bond. This would be a variant of the pinacol rearrangement.
Hey
Can you tell about ethyl shifts …
If there are two groups on carbon one methyl and second ethyl then which group will shift to carbonation
Good question. Ethyl (and other primary carbons) will migrate preferentially over methyl.
Hi! Why did another water molecule appear in the first example of a alkyl shift? I thought you already attached one molecule to the carbocation. Shouldn’t the “leftovers” be H+Cl-? I understand the extra hydrogen makes the other water molecule turn into hydronium ion but where did the other water come in the first place? Is this a water characteristic in reactions?
Thank you!
This would be with water as solvent (an ionizing solvent). Without the water, no substitution reaction would occur.
Why does the alkyl shift cause C2-C4 bond and not C2-C7?
Why rearrangement doesn’t occur at 1 butylcarbocation. If occurs what is the product when 1butene reacts with Hcl?
Once the tertiary carbocation forms, no rearrangement can occur that will make it more stable.
How do you decide between ring expansion,alkyl shift and hydride shift
is it necessary that such shifts occur only from the carbon atom adjacent to the positive charge??
Yes.
Please explain why the reaction of neopentane yields 1- chloro- 2,2-dimethyl propane when treated with chlorine molecule during free radical mechanism ?
why not 1,2 methyl shift to give more stable tertiary free radical as compared to less stable primary free radicle????
please reply.
1,2-shifts do not occur with radical substrates. Only carbocations.
Hi James,
Greetings.
How would you explain the methyl and ethyl carboxylate groups attached to the same carbon and only the ester group migrates as the 1,2 shift?
Is the migration via a radical or to the carbanion? I do not think that to the carbocation center. You said there are no 1,2-shifts in radicals.
Best,
Helena
It would help to see the specific example you’re thinking of, but it wouldn’t be a radical shift. There are two lone pairs on oxygen that could come down to help stabilize any positive charge buildup on the carbonyl carbon during the shift.
The 1,2-hydride shift occurs because of the hydrogen atom moving to the adjacent carbon with both of the electrons in the bond, according to what my teacher explained. He said that even alkyl groups can shift (though alkyls are much larger than hydrogen) since only the electron cloud is actually shifting.
Is it then possible to have any other shift that stabilizes the carbocation intermediate in a reaction, e.g. a -OH shift? You mentioned “allowed” rearrangement reactions, so what reactions are disallowed and why?
Thanks in advance!
Both reduction amd rearrangement occur in reaction…???
Replacing Cl with OH is not a reduction. Both are more electronegative than carbon.
I think in the very last reaction the attack by the water is shown at a wrong place.
no, i think it’s correct. When you expand you realize that the C that shifted is satisfied with 4 bonds C-H, C-CH2, C-CH3, C-C+. Whereas the C to which OH later gets attached has only 3 bonds C-CH2, C-CH3 and C-CH. Moreover if the OH had attached to the C you think it should’ve, it wouldn’t have enough free valence e- to another atom.
I hope I haven’t confused you.
Fixed, thank you Satyajit.
Why the attack of water molecule is at C-2 instead of C -3 carbocation.please explain.drklbajaj
It was an error. Thanks for correcting me Krishnan.