Conformations and Cycloalkanes
Bredts Rule (And Summary of Cycloalkanes)
Last updated: May 30th, 2026 |
Bredt’s Rule: Why Don’t Bridgehead Double Bonds Form?
This post is all about Bredt’s Rule (1924): double bonds cannot be placed at the bridgehead of a bridged ring system. We review the key points from our chapter on cycloalkanes, dive into Bredt’s rule, explain what’s going on in Bredt’s rule (spoiler: poor orbital overlap between p-orbitals) and then (Note 1) show that it’s actually more like “Bredt’s Strongly Worded Suggestion” since there are indeed some molecules which possess bridgehead olefins. And we also cover bridgehead amides.
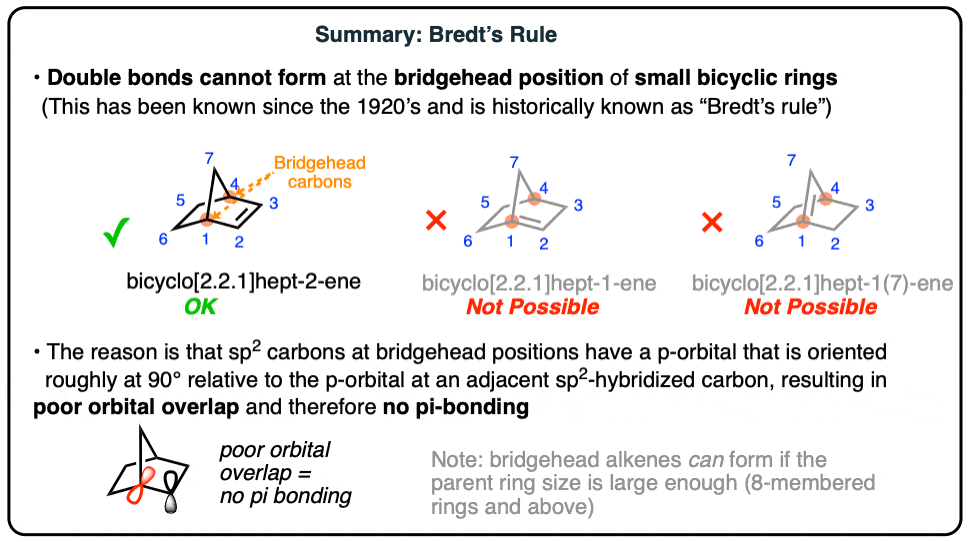
Table of Contents
- Cycloalkanes: What We’ve Learned So Far
- Bredt’s Rule: A Double Bond Cannot Be Placed At The Bridgehead Of A Bridged Ring System
- Bredt’s Rule Explained: Bridgehead Double Bonds Have Poor Orbital Overlap
- Summary: Bredt’s Rule
- Notes
- Quiz Yourself!
- (Advanced) References and Further Reading
1. What We’ve Learned So Far About Cycloalkanes
At the beginning of this series on cycloalkanes we saw that carbon’s ability to form rings leads to all kinds of interesting consequences that follow logically from the rules of structure and bonding in organic chemistry, but nevertheless would have been hard to predict from first principles. Among them, we’ve seen that:
- 3 and 4 membered rings have significant ring strain (a combination of “angle strain” and “torsional strain”)
- Rings of size <8 cannot turn inside-out, meaning that configurations of substituents relative to each other are locked. The simple way to say this is that it leads to the existence of cis and trans isomers for the disubstituted cases ( sometimes called “geometric isomers”) – e.g. cis– and trans- 1,2-dimethylcyclohexane.
- The most stable conformation of cyclohexane is the chair conformation in which all the groups along each C–C bond are staggered relative to each other. In the cyclohexane chair, there are two orientations of substituents on tetrahedral carbon: straight up/down relative to the ring (“axial”) and in the plane of the ring (“equatorial”).
- Cyclohexane chairs can undergo “flips” whereby all equatorial groups become axial and all axial groups become equatorial. This occurs rapidly at room temperature – so rapidly that in most cases at room temperature, the signals corresponding to each individual chair cannot be observed by our most useful tool (NMR spectroscopy) – instead, an average is observed. The activation energy for a chair flip is about 10 kcal/mol, since in order for a chair to “flip”, the molecule must pass through the strained “half-chair” conformation (about 10 kcal/mol higher in energy than the chair).
- Axial substituents lead to greater torsional strain than equatorial substituents, since they experience two additional “gauche” interactions with the hydrogens on the ring carbons located two bonds away. Another way to look at the same phenomenon is to think of the axial group interacting with each of the axial hydrogens (“diaxial” interactions). For methylcyclohexane, the conformation where methyl is axial is 1.70 kcal/mol more unstable than the conformation where the methyl is equatorial, a number referred to as the “A-value” for CH3. A-values have been measured for a large number of mono substituted cyclohexanes. In particular, the A-value for t-butyl is so high (>4.5 kcal/mol) that cyclohexane rings with a t-butyl substituent are essentially “locked” in the position where the t-butyl is equatorial.
- A-values are additive. With di- and trisubstituted cyclohexanes, we can use A-values to determine which chair conformation is most stable.
- The stereochemistry of fused rings can have a huge effect on the shape of the molecule. In cis-decalin, the molecule adopts a “tent” or “cup” shape where there is a concave and convex face. Trans-decalin is much more flat. Additionally, cis-decalin can undergo chair flips on each of its cyclohexanes, but trans-decalin is “locked” in position since a ring flip would lead to too much strain (essentially this would create the equivalent of a trans-double bond in a six membered ring, which is too unstable to form).
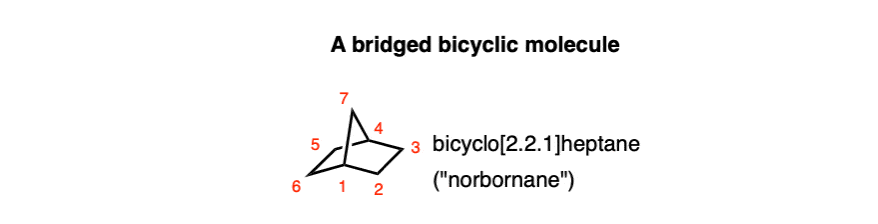
- “Bridged bicyclic” and “spiro” ring junctions are also possible. In bridged bicyclic molecules, the two bridgeheads are separated by “bridges” containing at least one carbon. In “spiro” fused molecules, the two rings are both joined at the same carbon.
Again, even if you are a genius, it would have been extremely difficult, if not impossible, for you to predict all this behaviour based solely on knowing the rules of chemical bonding. Many of these phenomena were first observed experimentally and the explanations provided post-hoc. That’s why I keep repeating the reminder that organic chemistry is very much an empirical science.
In this , the last post on this series on cycloalkanes, we’ll talk about a final surprising and interesting consequence of the fact that carbon can form rings. It goes like this:
2. A double bond cannot be placed at the bridgehead of a bridged ring system unless the rings are large enough.
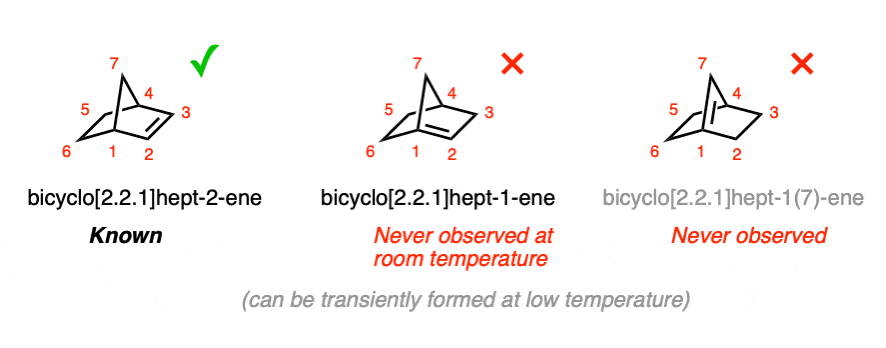
Let’s go through this. Imagine drawing a version of bicyclo[2.2.1]heptane (also called “norbornane”) with 1 double bond.
3 different constitutional isomers are possible. But only one has been observed at room temperature. (Very recently (2024), the transient existence of the middle bridgehead alkene was inferred by forming it at very low temperature and trapping it with a reactive partner)
Back in 1924, German chemist Julius Bredt was working on bicyclic molecules with these (and related) ring systems, and made the following generalization:
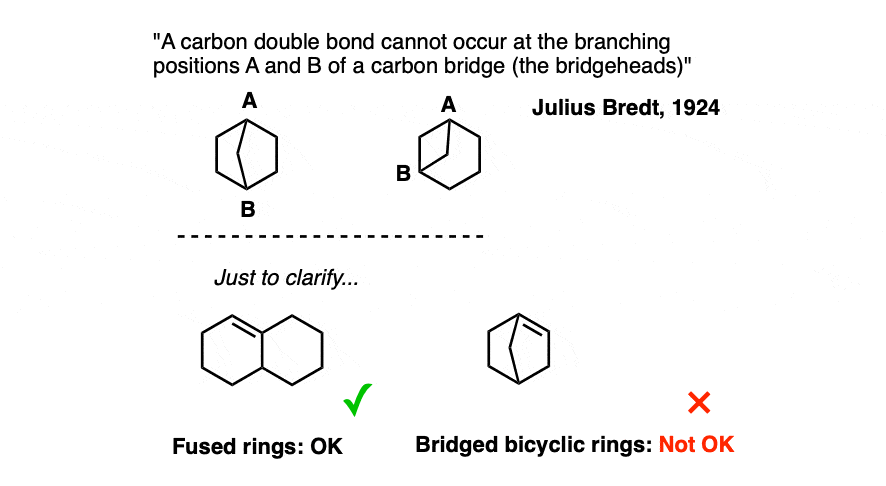
This observation came to be known as “Bredt’s rule“. Note that no deep explanation was offered at the time – merely the observation that these bridgehead double bonds do not form.
3. Bridgehead Double Bonds Have Poor Orbital Overlap
Looking at a model, and knowing what we know now about bonding, especially that of π bonds – can you think of a reason why bridgehead alkenes might be unstable?
Remember what is required in order for a π bond to form – we require overlap between the two adjacent p orbitals. In other words, they must be in the same plane, lined up like the plastic men on a foosball table.
Look closely at the bridgehead C-H bond in the model above. Note how it’s almost sticking straight out to the side of the molecule, especially with respect to the C-H bond on the adjacent carbon.
The normal geometry for a sp2 hybridized carbon is trigonal planar. However, due to the constraints placed on it by being part of multiple rings, the bridgehead carbon becomes “pyramidalized“, that is to say, somewhat like a pyramid (non-planar). In other words it has a very atypical shape for an sp2 hybridized carbon.
Since the model above doesn’t show p-orbitals, I’ve taken a photo of a model where the two p-orbitals are roughly indicated by the bridgehead C-H bond, and red loopy looking thingy on the adjacent carbon. Note that when we look along the C–C bond containing the bridgehead carbon, we see that the adjacent p orbitals are “staggered” with respect to each other. In other words there is very little orbital overlap. Poor overlap = poor bonding! [Note 2]
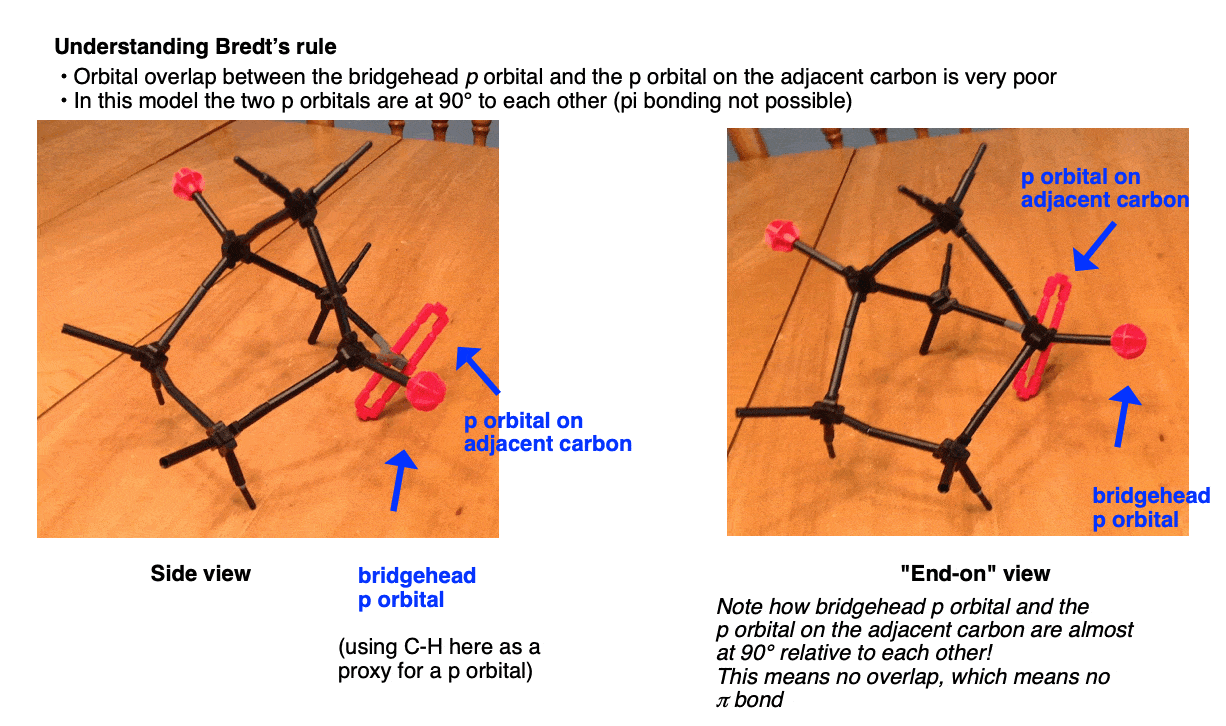
Summary: Bredt’s Rule
I didn’t show what the overlap in the other possible constitutional isomer would look like (the third case in the second diagram, above) but trust me when I say that the overlap is even worse.
So the bottom line for today is bridgehead double bonds are unstable due to poor orbital overlap.
Here endeth the lesson for the vast majority of readers. However, if you’re interested, you can continue reading to learn about how our understanding of Bredt’s rule has evolved since 1924.
Notes
Quiz Yourself!
Bredt’s rule has interesting consequences for many different reactions we’ll see in organic chemistry. This is a selection – you might not have seen these reactions before, and that’s OK! The key point is that the concept that we cover here comes up in a lot of different contexts, so it might be worth revisiting in the future. It’s a common source of “trick” questions!

Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.

Become a MOC member to see the clickable quiz with answers on the back.

Become a MOC member to see the clickable quiz with answers on the back.
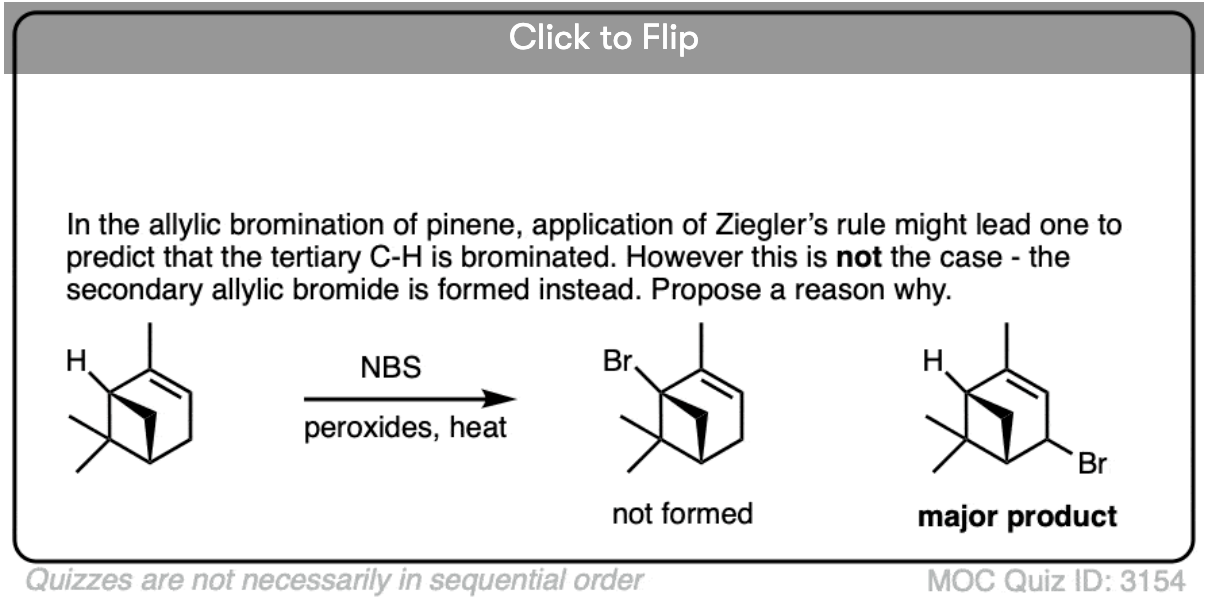
Become a MOC member to see the clickable quiz with answers on the back.
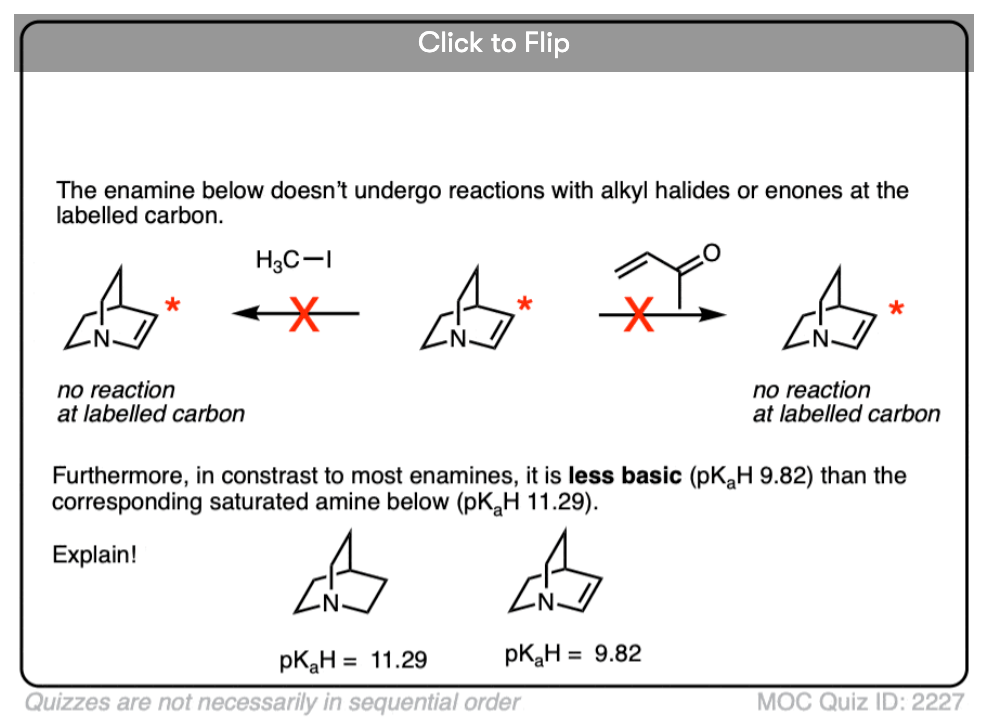
Become a MOC member to see the clickable quiz with answers on the back.
Exceptions To Bredt’s Rule, and Bridgehead Amides
[Nov 2024]. OK, turns out the parent alkene can be made, by inducing elimination of a silyl group and a halide through treatment with fluoride ino. Alas, as expected, the resulting bridgehead alkene is really unstable and can be trapped at very low temperature.
See: “A Solution to the Anti-Bredt Olefin Synthesis Problem”, by McDermott et. al. in Science, vol. 386 no. 6721. DOI: 10.1126/science.adq3519
Bredt, You’ve Got It Going On
Exceptions To Bredt’s Rule
In the intervening years the question of bridgehead double bonds has been studied in considerably more detail. The review by Shea (academic paywall) although more than 30 years old, is still a very useful primer on the topic. Of particular interest is that several natural products have been isolated that contain bridgehead double bonds, such as CP-225,917 (left) and Taxol (right)
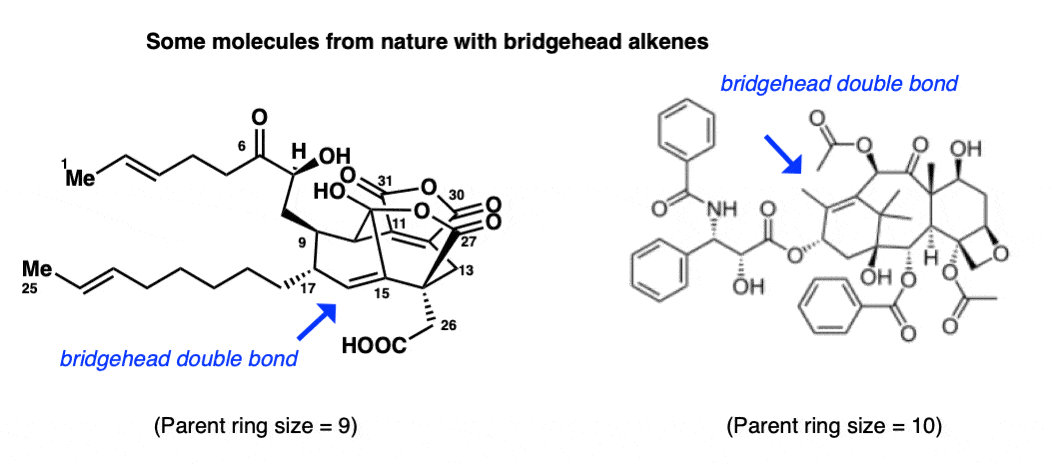
What gives? These molecules are clearly stable. Why might bridgehead double bonds be allowed in these cases, but not in the case of norbornene, above?
It has to do with the greater flexibility that accompanies larger ring sizes. It turns out that the stability of bridgehead double bonds roughly mirrors the stability of the largest trans-cycloalkene that contains the double bond.
Neither trans-cyclohexene nor trans-cycloheptene are stable enough to exist at room temperature (too much ring strain); nor have the bridgehead alkenes in a parent ring of 6 or 7 been observed as anything other than transient intermediates.
However, trans-cyclooctene is a stable molecule – and likewise, bicyclo[3.3.1]non-1-ene is a stable compound.. Observing bridgehead double bonds in molecules with parent ring sizes of 9 (CP-225,917) and 10 (Taxol) is therefore to be expected, since trans-cyclononene and trans cyclodecene are stable molecules, as are all the higher cycloalkenes.
Bridgehead Amides
If you are still reading this, you must be a nerd, so one last wrinkle.
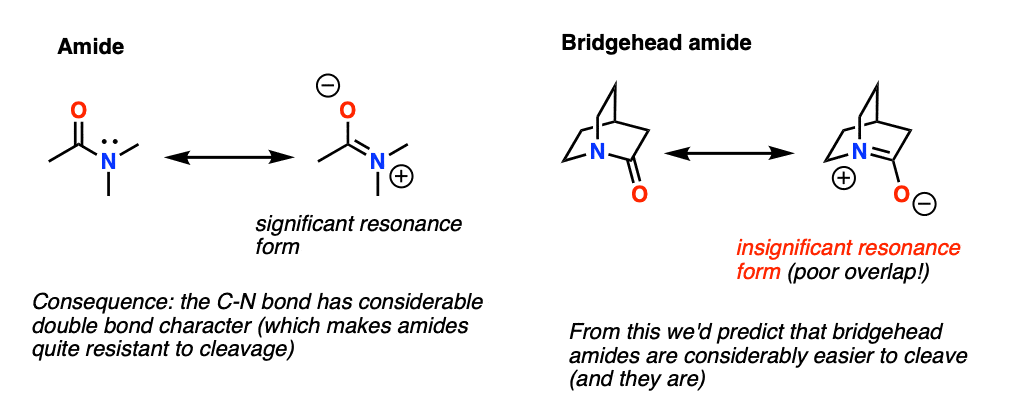
Amide bonds are generally difficult to break – certainly much more so than those of esters or acid chlorides. This is a good thing for us – strong peptide bonds make for stable proteins. One of the reasons is the considerable double-bond character between C and N as shown in the right-hand resonance form, a consequence of nitrogen’s considerable electron -donating ability.
What happens when the nitrogen is at a bridgehead, especially in a small ring system?
Needless to say the overlap between the lone pair on N and the carbonyl carbon is now extremely poor. This results in a considerably weaker C-N bond, one that is very easily cleaved by moderate nucleophiles.
In 2006 Brian Stoltz’ group at Caltech succeeded in isolating the HBF4 salt of the bridgehead amide above. Short and interesting paper.
Note 1: Another way of stating Bredt’s rule (once you learn about elimination reactions, is the following: “Elimination to give a double bond in a bridged bicyclic system always leads away from the bridgehead”
Bredt’s observations:
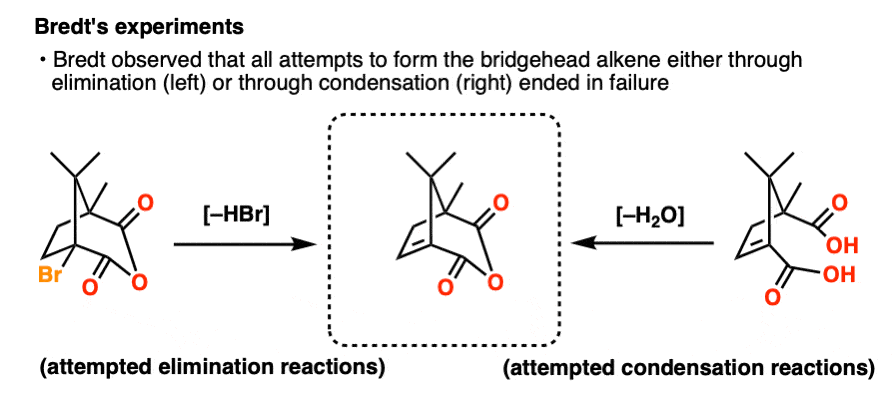
Note 2: In fact, double bonds of this type are predicted to have “diradical” character. Attempts to form bridgehead double bonds in rings of sizes 6 and 7 are often accompanied by phenomena such as dimerization and trapping of O2, which are clear indicators of radical intermediates.
(Advanced) References and Further Reading
- Über sterische Hinderung in Brückenringen (Bredtsche Regel) und über die meso‐trans‐Stellung in kondensierten Ringsystemen des Hexamethylens
J. Bredt
Justus Liebigs Annalen der Chemie 1924, 437 (1), 1-13
DOI: 10.1002/jlac.19244370102
The original paper by Bredt, noting the difficulty of synthesizing bicyclic compounds with a double bond at the bridgehead position. - Bredt’s Rule of Double Bonds in Atomic-Bridged-Ring Structures
Frank S. Fawcett
Chemical Reviews 1950, 47 (2), 219-274
DOI: 1021/cr60147a003
An old review from 1950 that compiles experimental observations to that date in support of Bredt’s Rule. - Bicyclo[3.3.1]non-1-ene
James A. Marshall and Hermann Faubl
Journal of the American Chemical Society 1967, 89 (23), 5965-5966
DOI: 1021/ja00999a049 - The Decarboxylation of β-Keto Acids. II. An Investigation of the Bredt Rule in Bicyclo[3.2.1]octane Systems
James P. Ferris and Nathan C. Miller
Journal of the American Chemical Society 1966 88 (15), 3522-3527
DOI: 10.1021/ja00967a011
A study of decarboxylation rates in bridged bicyclic keto acids, where the rate of decarboxylation is found to be strongly dependent on orbital overlap of the departing C-CO2H bond with the neighboring carbonyl. Good exam question material. - Bredt’s rule. Bicyclo[3.3.1]non-1-ene
John R. Wiseman
Journal of the American Chemical Society 1967, 89 (23), 5966-5968
DOI: 10.1021/ja00999a050
The above two back-to-back papers are on the same topic – the successful synthesis and isolation of the smallest compound with a bridgehead olefin. - Bredt’s rule. III. Synthesis and chemistry of bicyclo[3.3.1]non-1-ene
John R. Wiseman and Wayne A. Pletcher
Journal of the American Chemical Society 1970, 92 (4), 956-962
DOI: 10.1021/ja00707a035
This followup paper to Wiseman’s communication (Ref #4) provides more details on the synthesis and reactivity of the ‘Anti-Bredt’ olefin, bicyclo[3.3.1]non-1-ene. - Bredt Compounds and the Bredt Rule
Dr. Gert Köbrich
Angew. Chem. Int. Ed. 1973, 12 (6), 464-473
DOI: 10.1002/anie.197304641
As the structural basis for Bredt’s Rule became clear, it was evident that the prohibition against bridgehead double bonds would not be absolute. - Recent developments in the synthesis, structure and chemistry of bridgehead alkenes
Kenneth J. Shea
Tetrahedron 1980, 36 (12), 1683-1715
DOI: 1016/0040-4020(80)80067-6
This review summarizes work that took place after Fawcett’s review (Ref #2) and includes information on the successful synthesis of compounds with bridgehead alkenes. - Strained bridgehead double bonds
Philip M. Warner
Chemical Reviews 1989, 89 (5), 1067-1093
DOI: 1021/cr00095a007
A more recent review, this includes information on the successful synthesis of ‘anti-Bredt’ hydrocarbons. - Natural Products with Anti‐Bredt and Bridgehead Double Bonds
Jeffrey Y. W. Mak Dr. Rebecca H. Pouwer Assoc. Prof. Craig M. Williams
Angew. Chem. Int. Ed. 2014, 53 (50), 13664-13688
DOI: 10.1002/anie.201400932
This review covers the synthesis of natural products with bridgehead olefins. Compounds that have bridgehead double bonds and are otherwise stable are also known as ‘Anti-Bredt’ olefins, since they defy Bredt’s Rule. - Synthesis and structural analysis of 2-quinuclidonium tetrafluoroborate
Tani, K., Stoltz, B.
Nature 2006, 441, 731–734
DOI: 10.1038/nature04842
A landmark paper on the synthesis and unambiguous characterization (by X-ray spectroscopy) on potentially the smallest bridgehead amide. - Formation of Anti-Bredt Olefins from Bridgehead Carbene Precursors: A Computational Study
Michael Geise and Christopher M. Hadad
Journal of the American Chemical Society 2000, 122 (24), 5861-5865
DOI: 10.1021/ja000295g
This paper examines computationally whether ‘Anti-Bredt’ olefins can be formed by formation of the bridgehead carbene followed by rearrangement. In the conclusion, the paper states, “this study has shown that the equilibria for rearrangement of bridgehead carbenes to anti-Bredt olefins lie heavily on the side of the olefin. Also, the calculated intrinsic barriers to rearrangement are all easily accessible under most reaction conditions, with the largest barrier being 7.4 kcal/mol”. Now, all we need is experimental evidence! - Does 1‐Norbornene Exist?
R. Keese and E.‐P. Krebs
Angew. Chem. Int. Ed. 1972, 11 (6), 518-520
DOI: 10.1002/anie.197205181
1-norbornene, the smallest bicyclic bridgehead olefin which has been investigated experimentally, has not been directly observed or characterized. Instead it has been trapped as an adduct with furan, suggesting that it formed as an intermediate. - Evaluation and prediction of the stability of bridgehead olefins
Wilhelm F. Maier and Paul Von Rague Schleyer
Journal of the American Chemical Society 1981, 103 (8), 1891-1900
DOI: 10.1021/ja00398a003
This is a rather comprehensive computational study. Prof. Schleyer has calculated the strain energy and heat of formation of >50 bridgehead olefins, in order to determine trends based on structure. - A Solution to the Anti-Bredt Olefin Synthesis Problem
McDermott, L. et. al.
Science, 2024, 386, 6721
DOI: 10.1126/science.adq3519
At long last, a transient anti-Bredt olefin was formed through a fluoride-ion promoted elimination reaction and trapped at low temperature with various cycloaddition partners.
Hi Sir ,
Please explain that in a particular compound if Nitrogen is located at bridgehead position so is it able to donate it’s lone pair ? Is it basic ?
And if basic then you said sp2 carbon cannot be formed on bridgehead position so how and why nitrogen is able to donate it’s lone pair easily .
Source:(JEE MAIN 2025 4th April Evening Shift)
Please tell me sir
Hi – the issue is not that nitrogen can’t be on a bridgehead (it can – see compounds like quinuclidine and DABCO which are good bases) it’s that the lone pair on the nitrogen will not have good overlap with the p orbital on an adjacent carbon, which means that bridgehead amides will have very little C-N double bond character.
Does this article need an update now or is this Nature post impractical hype? “Chemists make ‘impossible’ molecules that break 100-year-old bonding rule” (https://www.nature.com/articles/d41586-024-03538-4)
McDermott, L. et al. Science 386, eadq3519 (2024). https://doi.org/10.1126%2Fscience.adq3519
Chan, T. H. & Massuda, D. J. Am. Chem. Soc 99, 936–937(1977). https://doi.org/10.1021%2Fja00445a042
Good question. I don’t think the new paper changes things all that much, but it’s a good addition to the literature.
I think the message from the new paper is that and olefins do form, but they just happen to be very unstable at low temperature and can be trapped.
Kind of like how cyclobutadiene can be formed at very low (35 K) temperatures but spontaneously dimerizes.
It is really helpful. Thanks a lot sir for such an awesome and detailed explanation :)
*carbonyl carbon. (under bridgehead amides) :)
Fixed. Thanks Pragna!
SN2 is possible at bridge head carbon or not?
Absolutely not! No backside attack possible.
What’s the name of the compound the one in bridgehead amide and in note1? Thanks
2-quinuclidone, or 1-azabicyclo[2.2.2]octan-2-one
Thanks, funny quote in between.
I have a doubt. Can carbanion be formed at bridgehead position of [2.2.1]bicyclo heptane?
Not easily. If there is a bromine at the bridgehead position, you could reduce it off with sodium metal, but the resulting anion will not be very stable.
Just curious but what if we compared the stability of bicyclo[2.2.2]octane vs bicyclo[2.2.2]oct-2-ene? Which would be more thermodynamically stable?