Bonding, Structure, and Resonance
In Summary: How To Evaluate Resonance Structures
Last updated: May 28th, 2026 |
The Four Key Factors In Evaluating Resonance Structures
Not all resonance forms are of equal importance. So how do we evaluate how “important” each resonance structure is?
As we’ve seen in previous articles, four key factors that determine the importance of resonance structures in organic chemistry are:
- Rule #1: Minimize charges
- Rule #2: Full octets are favored
- Rule #3: How stable are the negative charges?
- Rule #4: How stable are the positive charges?
Today, let’s summarize everything we’ve learned about resonance structures in this unit.
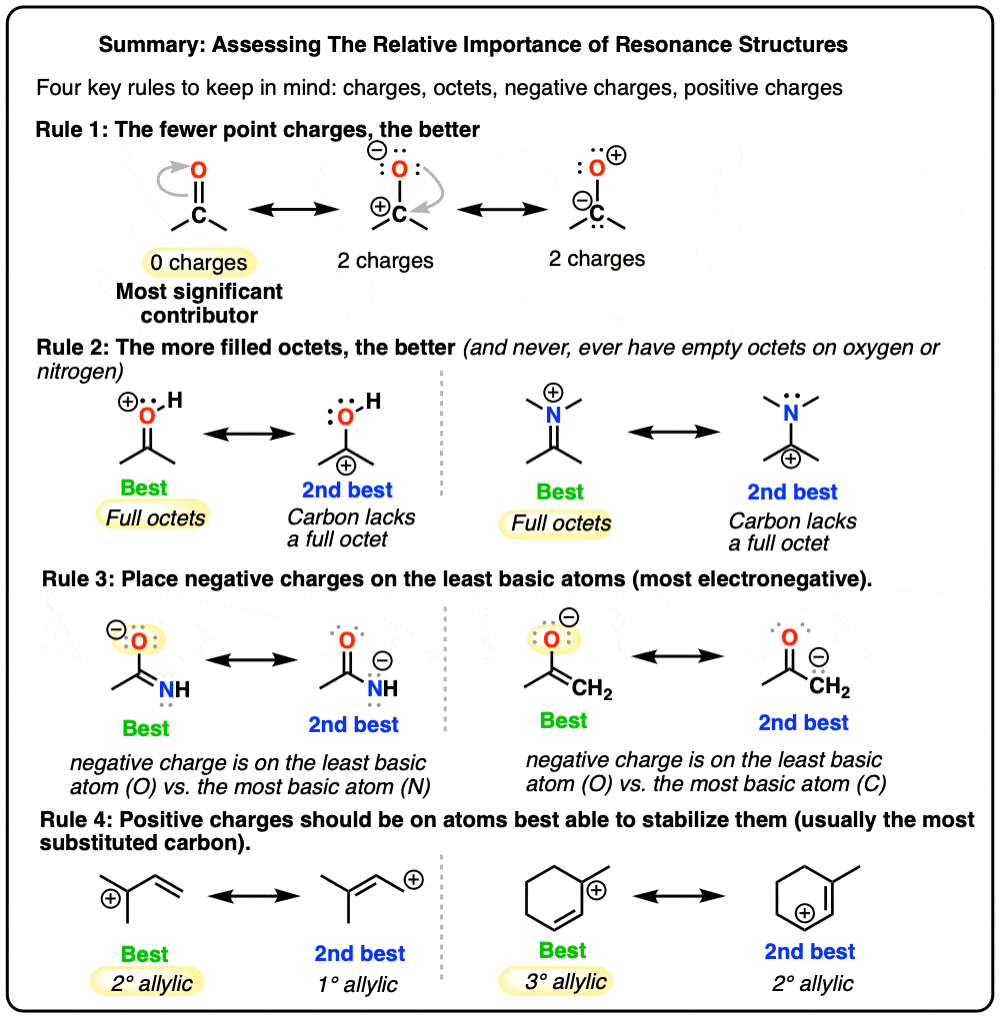
Table of Contents
- Recall The Three “Legal” Electron-Pushing Arrow “Moves” Used For Interconverting Resonance Structures
- Remember That Resonance Structures Are Not In Equilibrium With Each Other – They Represent Contributions To An Overall Resonance “Hybrid”
- Not All Resonance Forms Are Of Equal Significance. So How Do We Evaluate How “Important” Each One Is?
- Rule #1: Neutral Resonance Structures Are More “Important” Than Charged Resonance Structures
- Rule #2: Full Octets Are Preferable To Empty Octets (And Never, Ever Have Empty Octets On Oxygen or Nitrogen!)
- Rule #3: Place Negative Charges On The Atom Best Able To Stabilize It (i.e. The Least Basic Atom)
- Rule #4: Place Any Empty Octets On the Atoms Best Able To Stabilize Them (i.e. Carbon And Not Oxygen Or Nitrogen)
- An Application Of Resonance: “Pi Donation”
- A Second Application Of Resonance: “Pi Acceptors”
- Notes
- Quiz Yourself! (On Evaluating Resonance Structures)
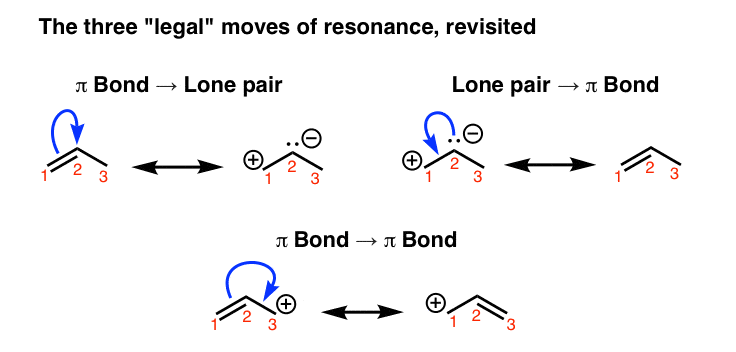
1. Recall The Three “Legal” Electron-Pushing Arrow “Moves” Used For Interconverting Resonance Structures
After all these posts about resonance, I thought it would be good to have a post summarizing what’s been discussed so far.
One of the key skills in analyzing the reactivity of a molecule is to be able to figure out where the electrons are.
As I wrote here, if we’re dealing with single bonds, it’s a relatively straightforward matter of figuring out the differences in electronegativities.
However if multiple bonds (π bonds) are present, then we start to run into a little problem: there can be multiple ways to distribute electrons on the same molecule (i.e. different resonance forms). Therefore, in order to understand electron density on a molecule where pi bonds are present, we must first understand the importance of its various resonance forms.
How can we “find” resonance forms for a given molecule? It’s possible to do it through trial-and-error, but one surefire way is to do so is to apply the curved arrow formalism, which is a way of depicting the “movement” of electrons.
There are three “legal” ways to move electrons using curved arrows: from pi bond to lone pair, from lone pair to pi bond, and from pi bond to pi bond:
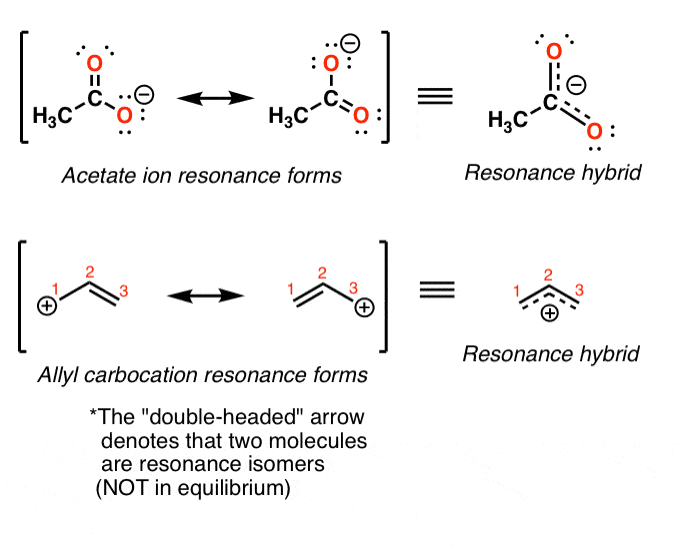
2. Remember That Resonance Structures Are Not In Equilibrium With Each Other – They Represent Contributions To An Overall Resonance “Hybrid”
Here’s an important point about resonance forms. It is tempting (and very wrong!) to think that these resonance forms are in “equilibrium” between each other. Avoid this common mistake!
Instead, the “true” state of the molecule will be a “hybrid” of these resonance forms.
For example in the acetate and allyl cation examples below, the “true” structure of the molecule is represented through a 50:50 combination of the two resonance forms.
3. Not All Resonance Forms Are Of Equal Significance. So How Do We Evaluate How “Important” Each One Is?
In the case of the acetate ion and the allyl cation, both resonance forms are equal in energy, so the “hybrid” is a 1:1 mixture of the two. However, this is only rarely the case.
Take the ketone below (acetone, or “propanone”) for which we can draw 3 different resonance forms.
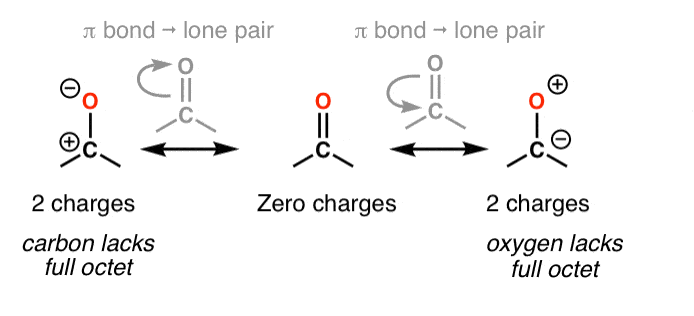
In cases like these, how do we evaluate the relative importance of each resonance form?
4. Rule #1: Neutral Resonance Structures Are More “Important” Than Charged Resonance Structures
Resonance forms become less significant as the number of charges are increased (see earlier post).
For example, in the ketone above, the resonance form with zero formal charges will be the most significant.
How do we know? We can measure the physical properties of the molecule (e.g. boiling points, solvent properties, conductivity) and see if it’s more consistent with a charged species or a neutral compound.
All the physical properties of propanone (acetone) are consistent with it being a (mostly) neutral molecule. For example acetone has a boiling point of 56°C, significantly lower than water, and a freezing point of –95°C. It doesn’t dissolve charged species (like NaCl) nearly as well as water does. And it isn’t a particularly good conductor of electricity.
That isn’t to say that the “second-best” resonance form doesn’t play some role.
Acetone is much higher-boiling than butane (–1°C) which has a similar molecular weight due to the dipole-dipole Van der Waals attractive forces, and as we’ll see later, the “second-best” resonance form can yield an important clue as to the reactivity of a molecule.
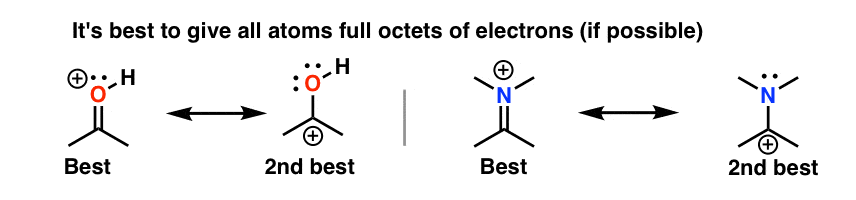
5. Rule #2: Full Octets Are Preferable To Empty Octets (And Never, Ever Have Empty Octets On Oxygen or Nitrogen!)
Resonance forms where all atoms have full octets will be more significant than resonance forms where atom(s) lack a full octet. Importantly, it’s a good general rule never to place less than a full octet on nitrogen or oxygen, as in the acetone example (above right). Since these atoms are highly electronegative, these resonance forms are extremely unstable and will be insignificant.
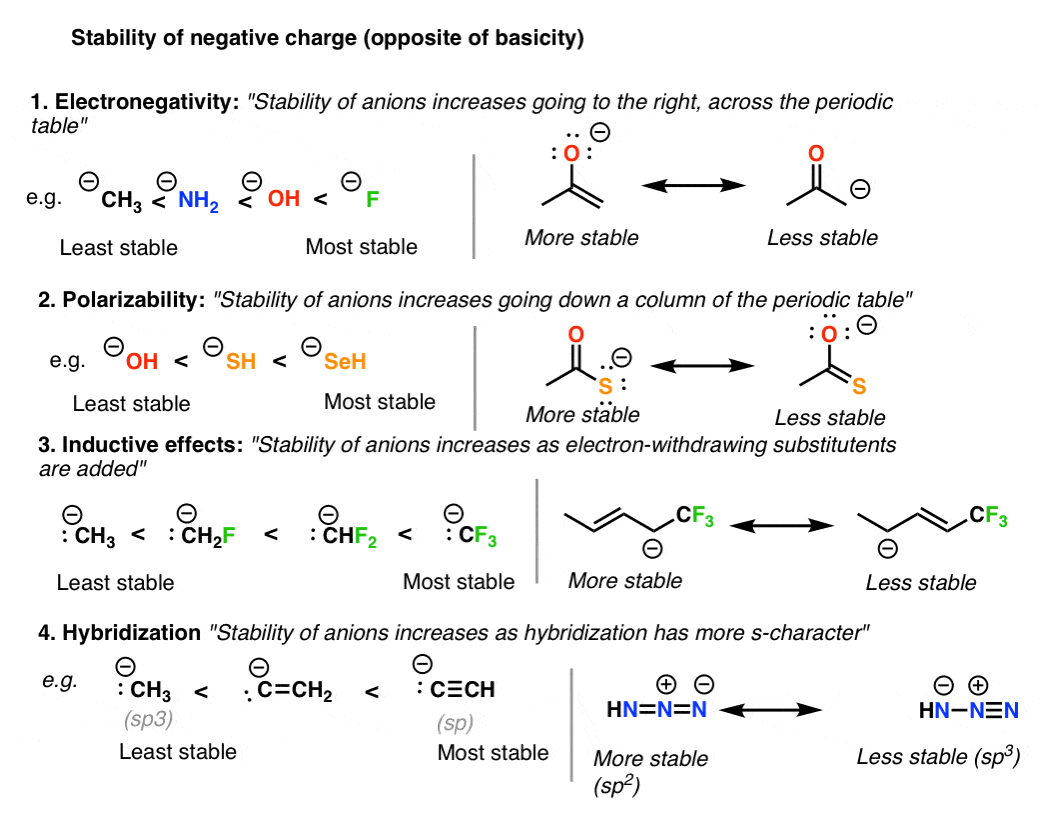
6. Rule #3: Place Negative Charges On The Atom Best Able To Stabilize It (i.e. The Least Basic Atom)
Given that neutral resonance structures are preferred overall, when a resonance structure absolutely must bear a negative charge somewhere, place it on the atom best able to stabilize that charge. Since, in essence, “basicity is the opposite of stability”, this is the same as saying, “put the negative charge on the least basic atom”.
The good news here is that if you understand the factors that affect acidity, you also understand the factors which stabilize negative charge.
There are four main trends to consider here:
- Electronegativity: across a row of the periodic table, negative charge becomes more stable as electronegativity is increased.
- Polarizability: down a column of the periodic table, negative charge becomes more stable as polarizability increases
- Electron withdrawing groups stabilize negative charge through inductive effects.
- Hybridization: negative charge becomes more stable as the s-character of the atom is increased. sp (most stable) > sp2 > sp3 (least stable
Note again that stability is the opposite of basicity.
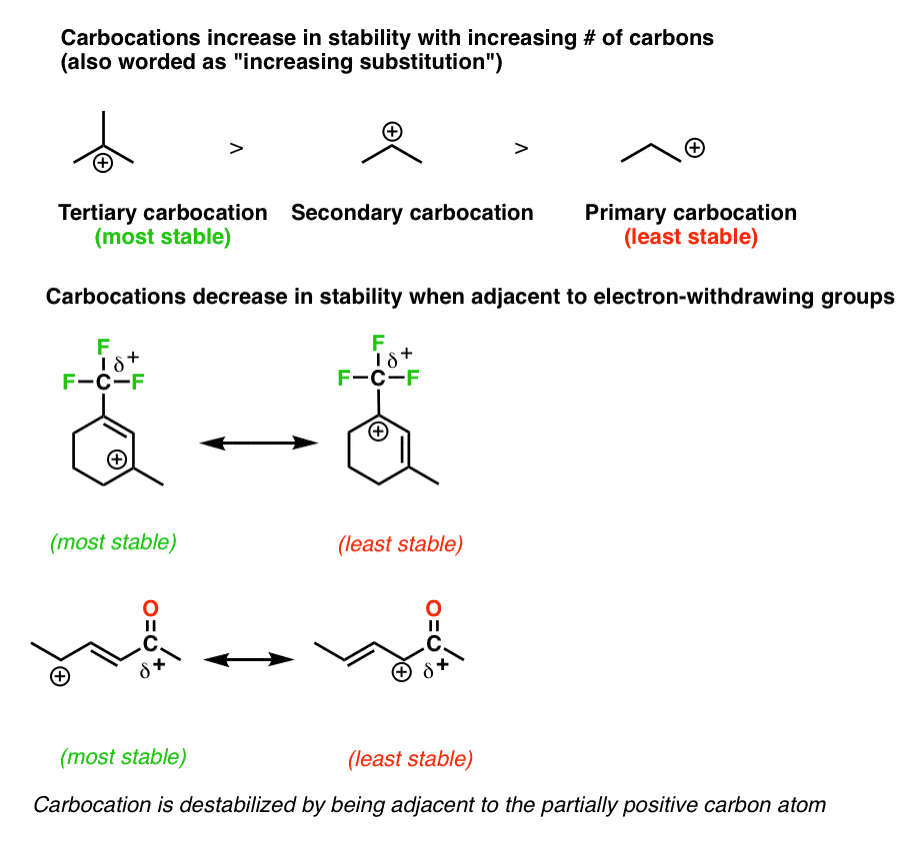
7. Rule #4: Place Any Empty Octets On the Atoms Best Able To Stabilize Them (i.e. Carbon And Not Oxygen Or Nitrogen)
As we said above, full octets are best. However, when dealing with a resonance structure where there absolutely must be an atom with less than a full octet, then follow these principles:
- Place the empty octet on carbon, never oxygen or nitrogen
- Place place the empty octet on the most substituted carbon (remember carbocation stability)
- Avoid placing positive charge adjacent to electron withdrawing groups if possible
- Place positive charge preferentially on alkyl carbocations as opposed to alkenyl or (especially) alkynyl carbons.
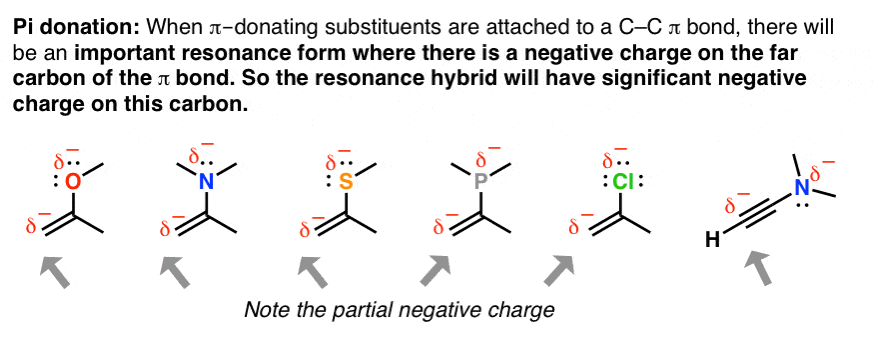
8. An Application Of Resonance: “Pi Donation”
When double bonds are connected to an atom with a lone pair of electrons, the molecule will have a significant resonance form where there is negative charge on the adjacent carbon due to a phenomenon called, “pi donation“. This becomes particularly important once you start learning about reactions of pi bonds.
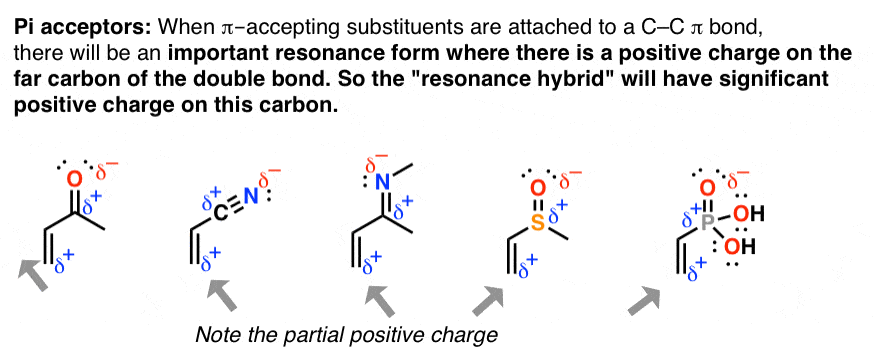
9. A Second Application Of Resonance: “Pi Acceptors”
When double bonds are connected to a polarized π bond, the molecule will have a significant resonance form where there is positive charge on the adjacent carbon. This phenomenon is known as “pi-accepting” behavior, and these groups are known as “pi acceptors“.
For now, that does it for a summary of the important themes in resonance. Next stop (after a post about some common mistakes) will be to apply these principles to chemical reactivity.
Next Post: Common Mistakes – How NOT to Draw Resonance Curved Arrows
Notes
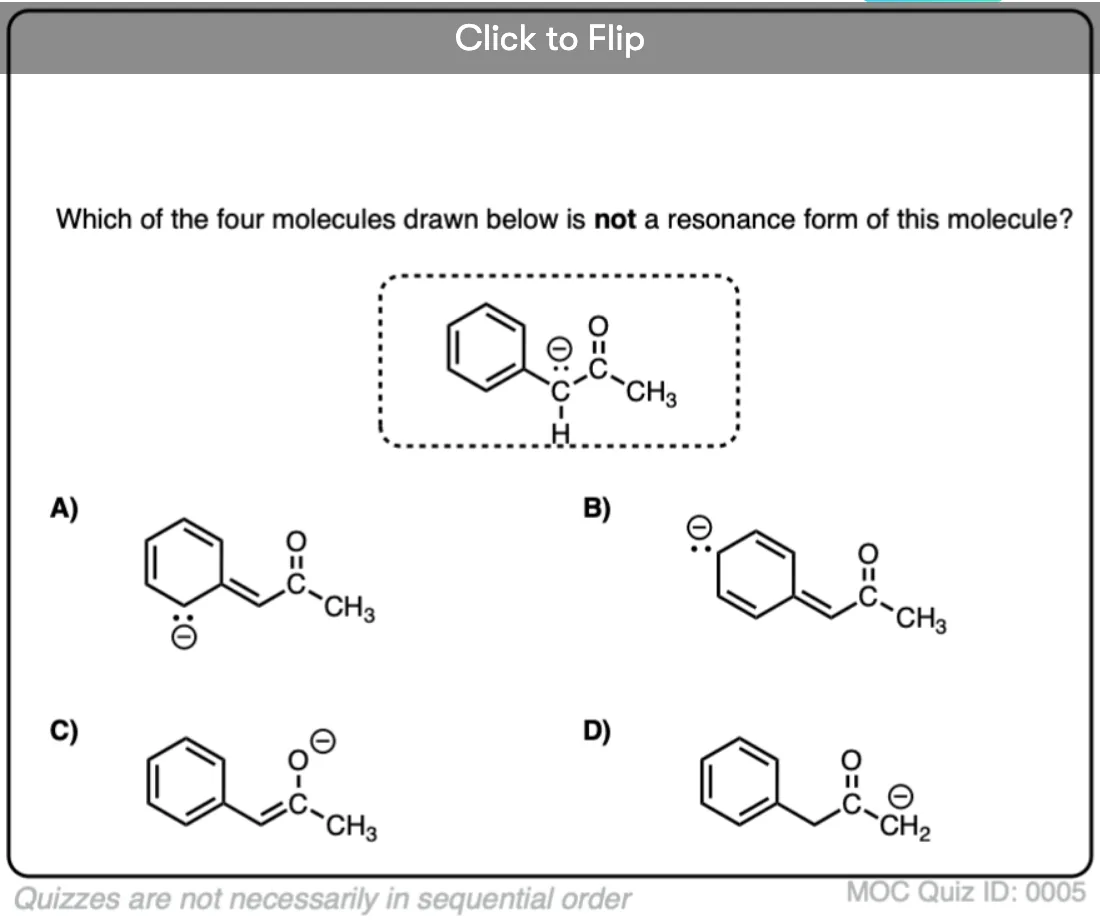
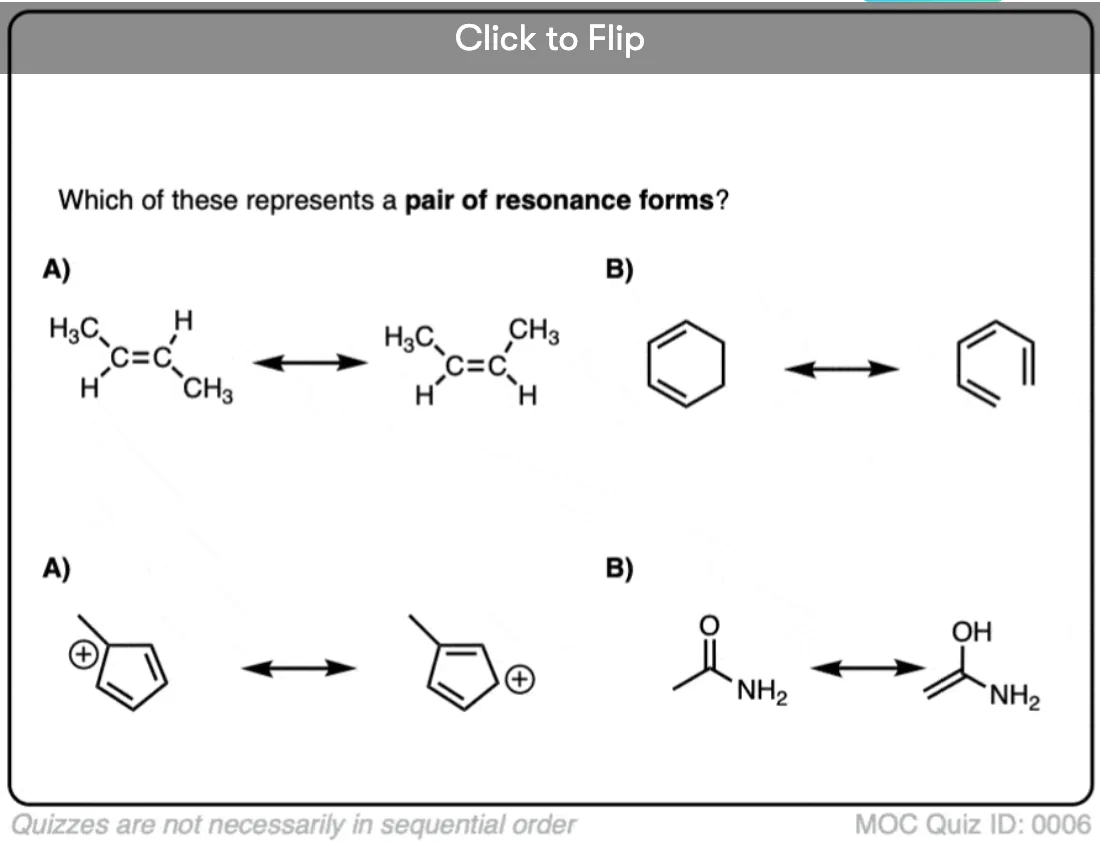
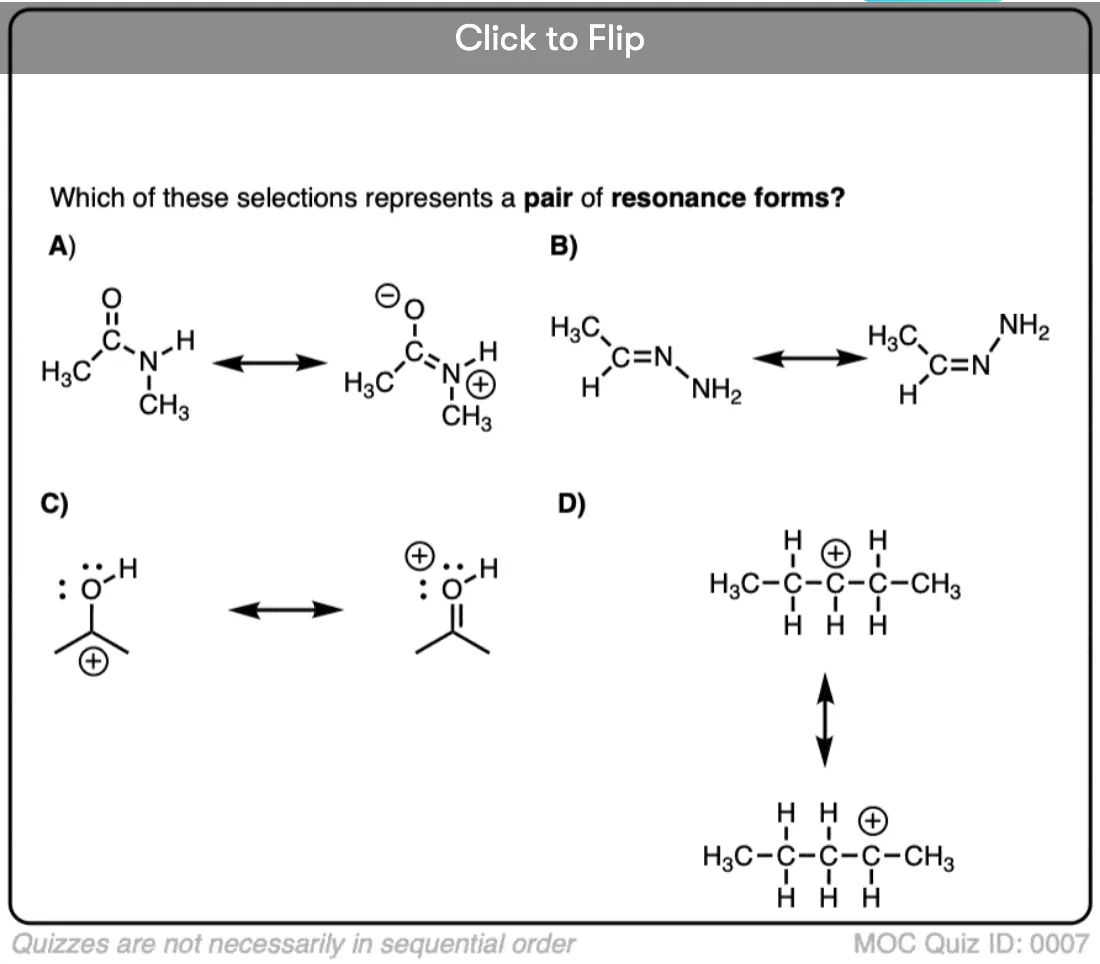
Quiz Yourself on Resonance Structures
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
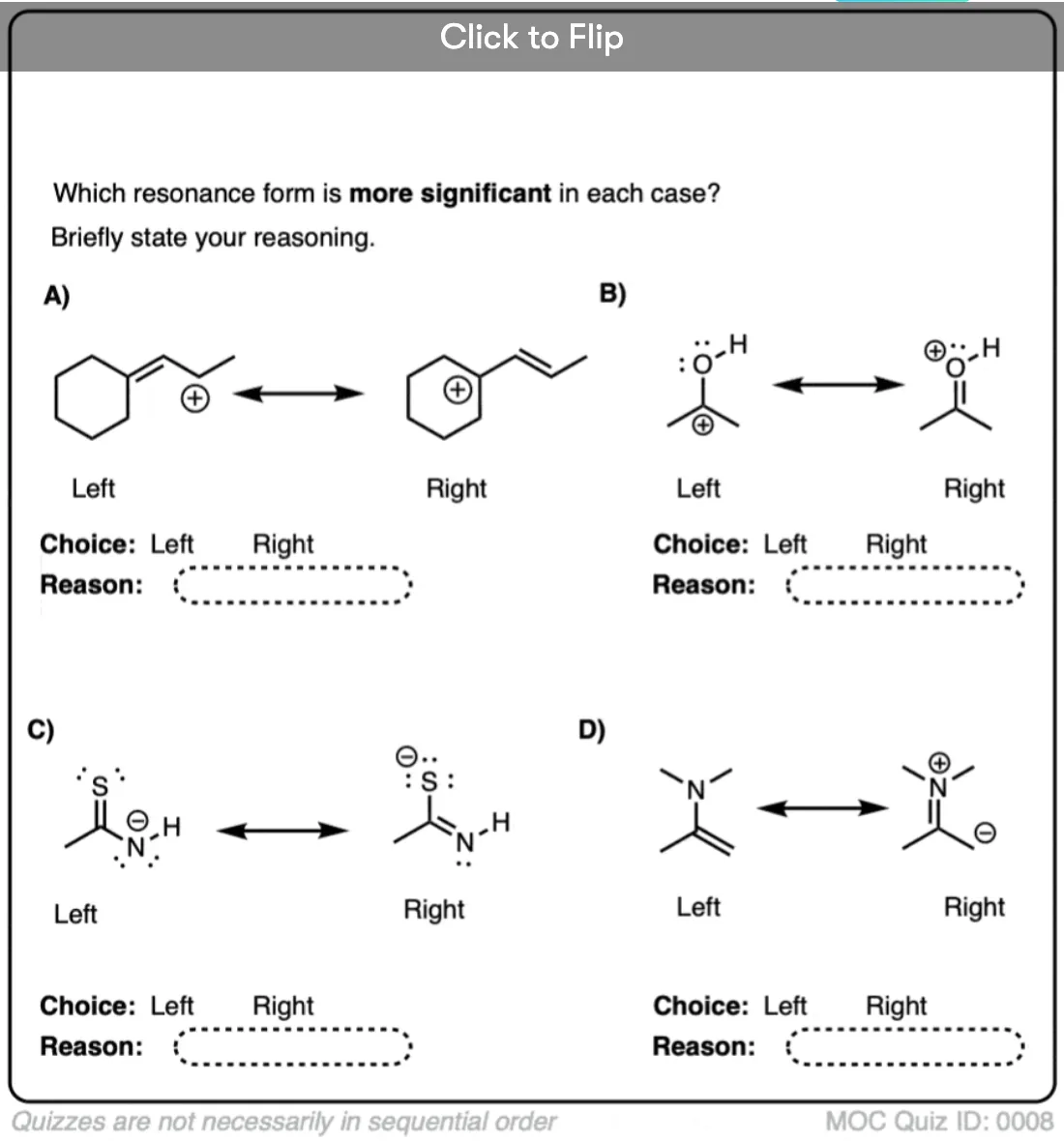
Become a MOC member to see the clickable quiz with answers on the back.
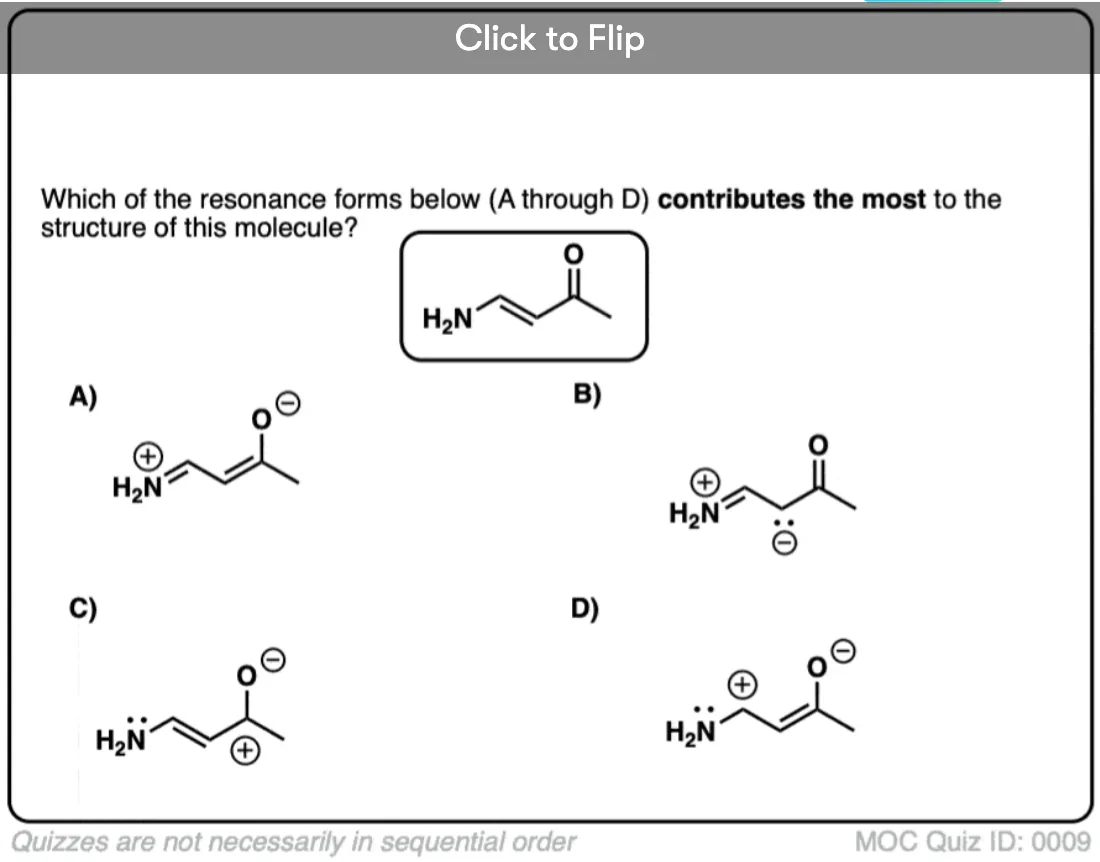
Become a MOC member to see the clickable quiz with answers on the back.
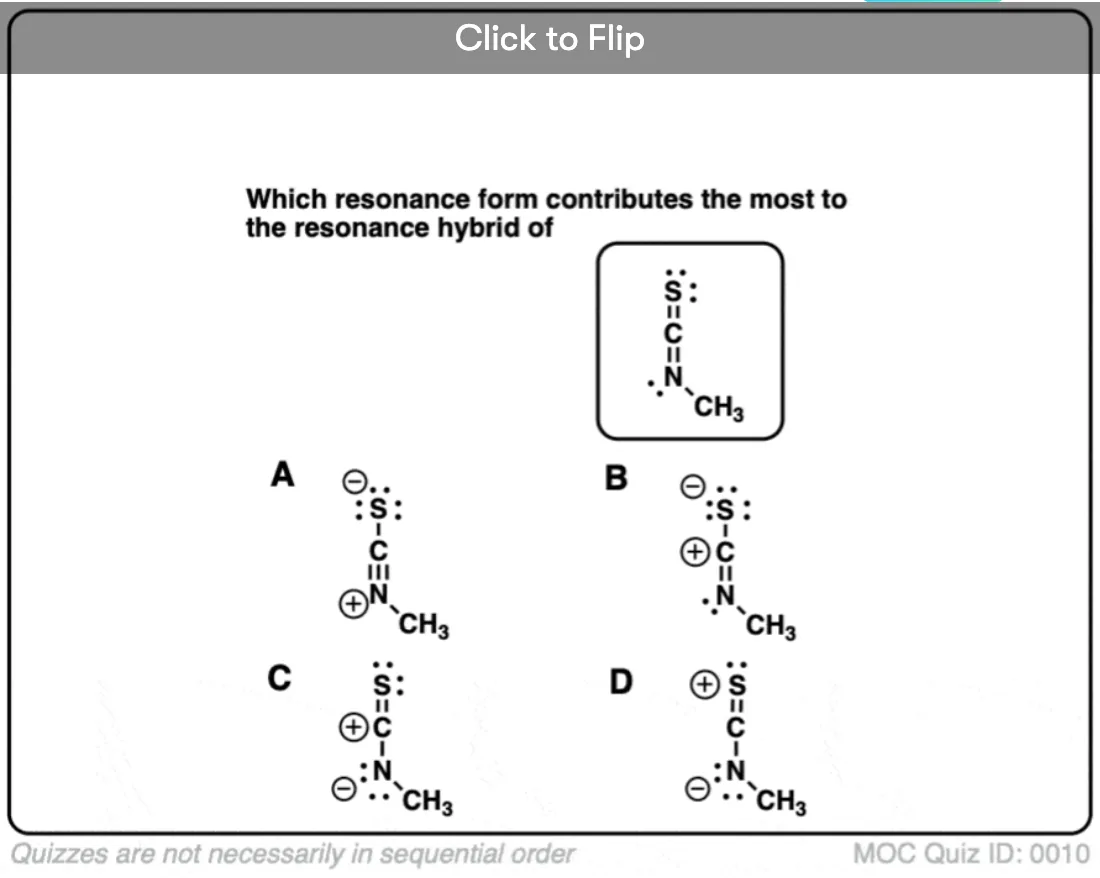
Become a MOC member to see the clickable quiz with answers on the back.
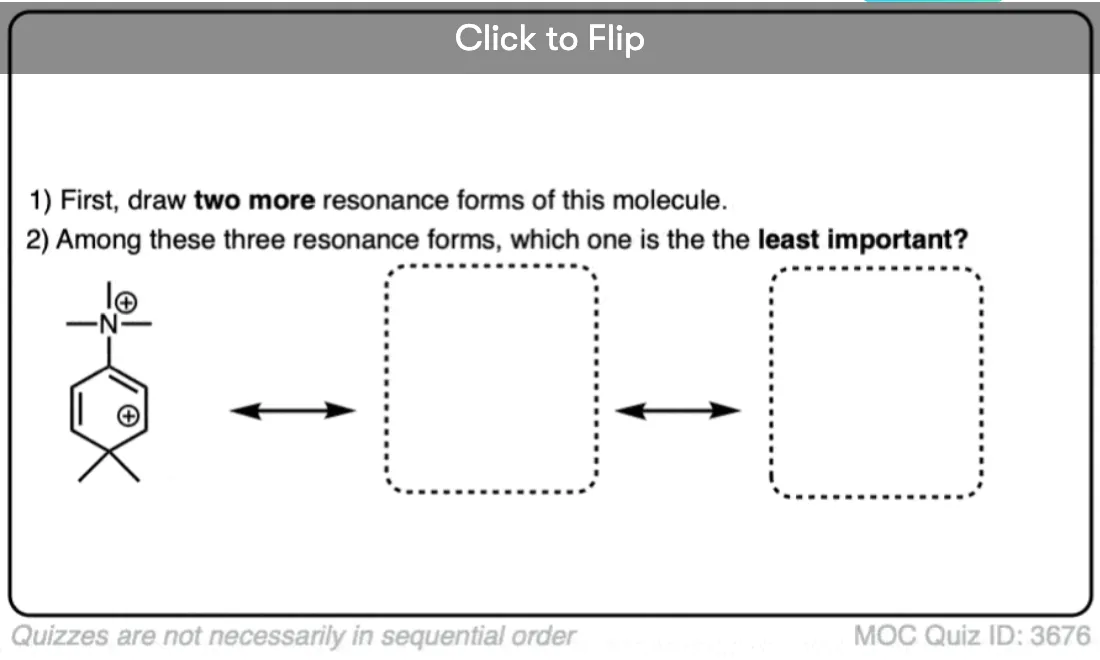
Become a MOC member to see the clickable quiz with answers on the back.
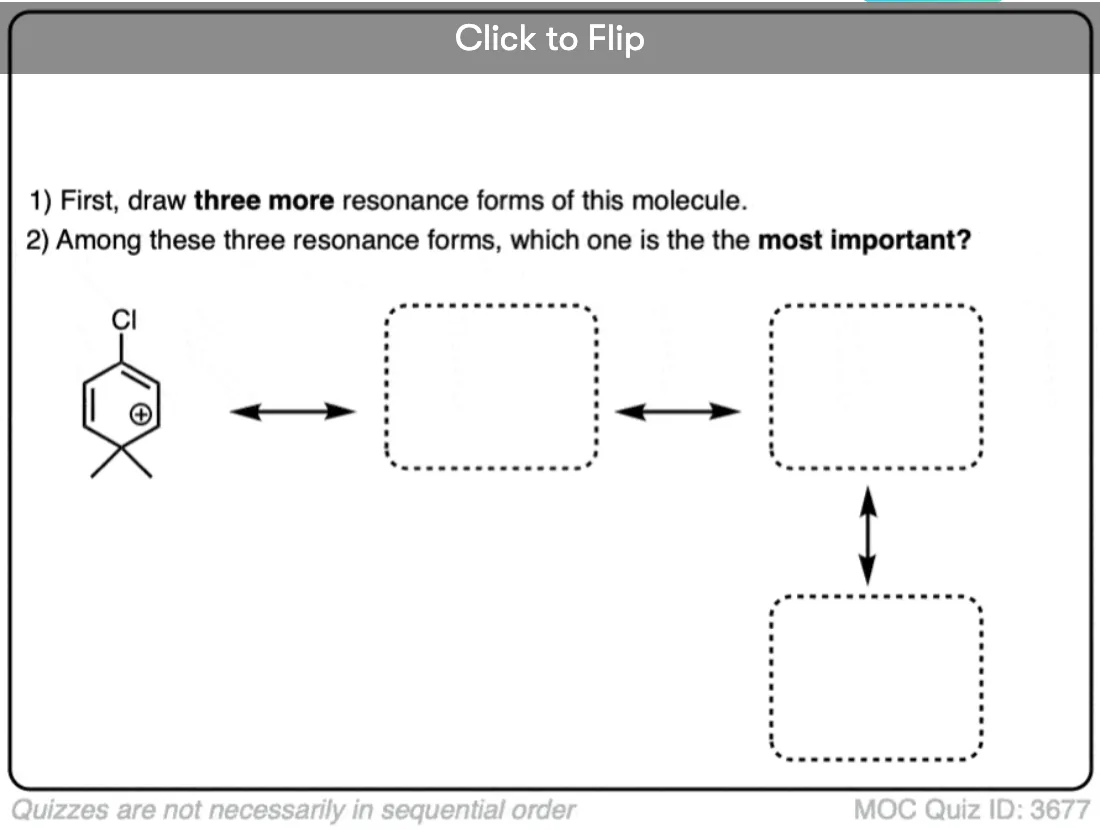
Become a MOC member to see the clickable quiz with answers on the back.
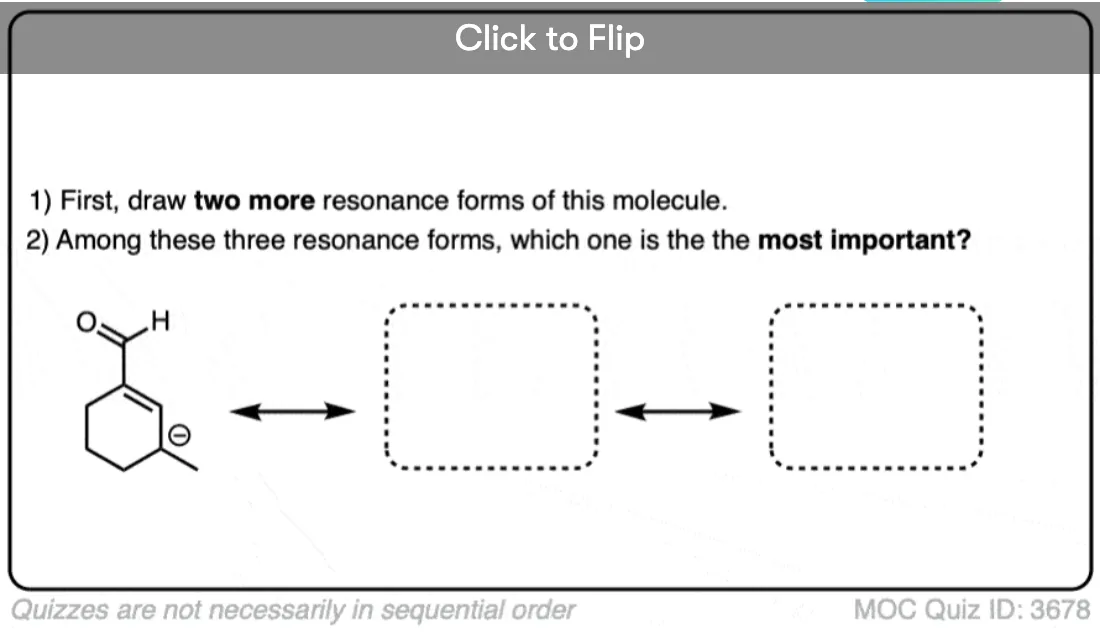
Become a MOC member to see the clickable quiz with answers on the back.
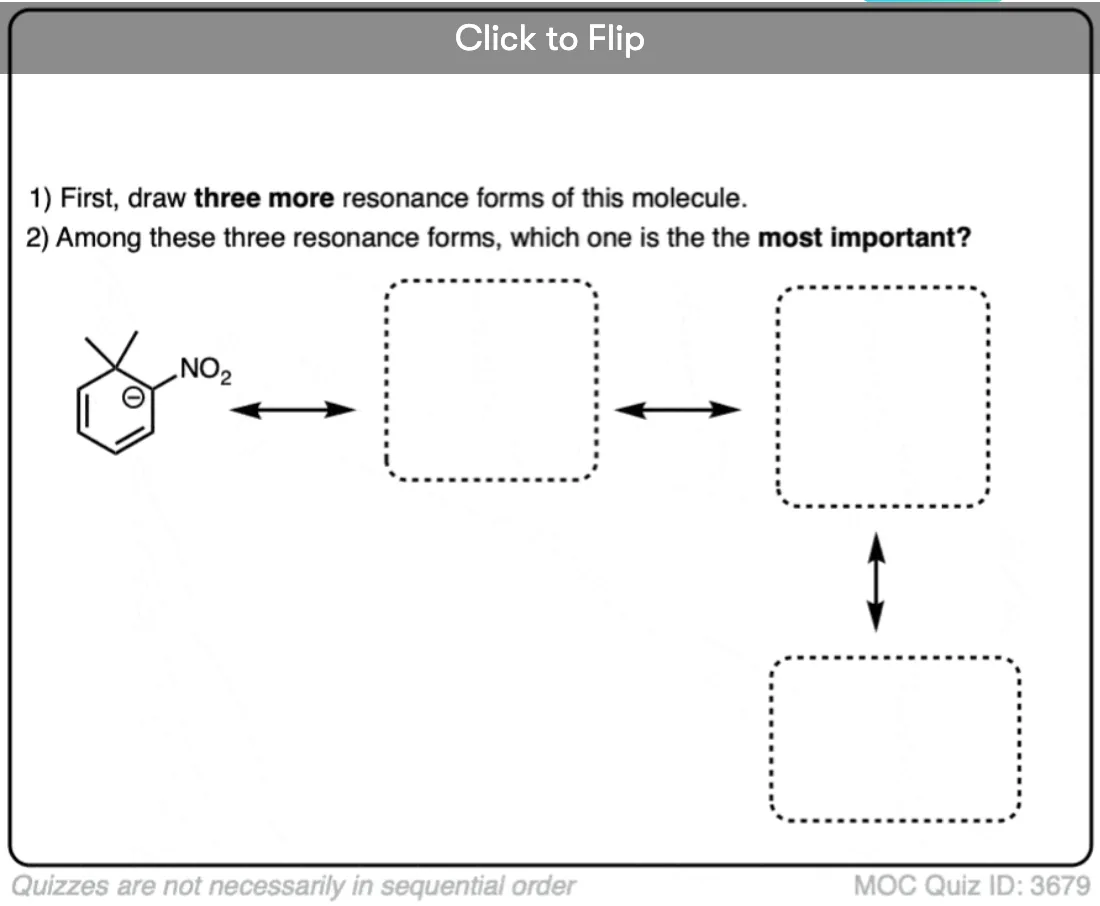
Become a MOC member to see the clickable quiz with answers on the back.
In the first infographic, Rule #4, there’s a typo. The infographic states that 2° allylic is the most stable, but it actually should be 3° allylic.
Great spot! fixed! let me know if you see any more!
I have understand this but I have a very small doubt that
What is Resonance why do we make resonating structures ?
We make resonance structures because it is otherwise very difficult to simply show the electron density in structures with partial bonds.
What about charge separation how does that factor stack up against other factors you have mentioned???
I see absolutely no case for charge separation being a factor. Opposite charges attract anyway.
General chemistry teaches the evils of charge separation, but far, far overstates it. Charge separation is awful in the gas-phase, but in polar/complexing solvent, it is not nearly as bad as is suggested. The bigger, overriding issue is not the charge separation but the lack of the octet that normally accompanies it. I’d take charge separation with octets over no-charge separation without octets (take that “expanded octet” nonsense!).
But even with lack of octets, we know that charge separation is not bad when you have polar bonds. The carbonyl is a very polar bond. Hence, we can say that the polarized resonance structure is important.
Thanks for weighing in Paul!
Please tell me about the resonance effect of NO group on benzene ring.
NO is a sigma acceptor but a pi donor. It is an ortho-para director, similar to halogens. https://www.masterorganicchemistry.com/2018/03/05/why-are-halogens-ortho-para-directors/
does low stable molecule contribute to resonance
I have problems when i try to find out the hybridisation in the structures which show the phenomenon of resonance.Can’t we find it out using steric no.? Also I had problems regarding how will the electron density be represented in the p orbital or pi orbital.I just get confused when I try to fin the relation between vbt, mot and resonance
Got it!a nice treatment on my doubts on http://web1.uct.usm.maine.edu/~newton/Chy251_253/Lectures/Aromaticity/AromaticityFS.html
Also this pdf helps:http://www.chem.ucla.edu/harding/tutorials/aromaticity.pdf
BTW It’s an exceptional site you have got there and It’s helped me even more than my tuition teacher!
Yes – UCLA is a great site.
uhhhhh……
I actually mentioned your site as great!!!
Thanks James. In your answer, you touch upon something we often take for granted, but which I will call “implicit semantics”. If the participants in the “shared space” all agree that they know an anionic centre has an “invisible” lone pair, then of course it is OK to start the arrow at the charge and dispense with drawing the lone pair. But I remind of the fascinating story of how benzene has been represented over the years. Faraday who discovered it, attributed no structure. Loschmidt controversially represented it as a circle, and Kekule gets the credit for the final form. But after that chemists got lazy. They dispensed with Kekule’s two forms, reducing them not only to one (the other being now implicit) but they also eliminated the three double bonds, making these also implicit. For perhaps 70 years, it became normal to draw a benzene ring as a pure hexagon, and to leave the unsaturation as implicit. Text books up to around 1950 appear to be describing cyclohexane, whereas in fact we might conclude that they are actually referring to benzene itself..
Benzene is also associated with another form of implicit meaning. Robinson around 1925 coined the term aromatic sextet, and mesmerised by this, anointed naphthalene with two rings (after all, the two rings are related by a plane of symmetry). The use of two such inner circles for naphthalene implies two sextets, or 12 π electrons. Of course we know that two such sextet rings actually implies 10 electrons! See what happens when you take short cuts!
Thanks for the historical perspective. I was unaware about the cyclohexane example. And the doubly circled rings for naphthalene is definitely something to avoid.
[I made a longer comment previously but decided to delete it. There’s too many misunderstandings possible when communicating through blog comments].
The very first diagram above shows some curved arrow formalism (with a reference to Wikipedia). The second example shows a lone pair to π-bond resonance. It might seem trivial, but it does matter where one shows an arrow “starting” from; in this case a negative charge on a carbon. Might I suggest that a slightly more consistent approach is to start arrows not so much at charges, but at the associated lone pair (also shown in your diagram). One can locate the centroids of electron (pairs), and therefore in principle one can define coordinates for the start of an arrow (and also by the same principle, coordinates for the destination of the arrow). It is not however possible to define coordinates for a “charge” (other than placing it at the centre of the atom itself, which is hardly a very electronic thing to do!).
I have touched upon this theme here where you might want to contemplate the arrows for the reverse of the three reactions shown. Also here.
Thanks for the input Henry. You’ve pointed out a common dilemma. It is indeed more consistent to show arrows starting at lone pairs instead of charges. There are costs and benefits to each approach. The benefit of drawing all the lone pairs in is that there is no ambiguity about the location of the electrons and it is more proper to do so. There is, however, a cost both in the extra clutter of the drawings and also in the extra time involved in drawing in these lone pairs. For that reason it is *extraordinarily* common for students and instructors to omit drawing lone pairs when a formal negative charge *and* a lone pair is present on the same atom, and simply draw the curved arrow as coming from the charge (in the absence of a formal negative charge, a corresponding lone pair *must* be drawn in however). I choose to do this as well. So long as this caveat is understood (i.e. that the negative charge “stands in” for a lone pair on the atom) in my experience there has been little confusion on this point. However it is not without regret that some information content is lost.
Fixed the diagram. You’re right, it was a little sloppy coming from the charge instead of the lone pair. Thanks for the correction.
Maybe I missed it in earlier blogs, but what exactly is resonance? Do you think of it as superposition, where several different structures exist at the same instant? If so, then you cannot think of any of your structures as single molecules. But if you think in terms of single molecules, then only one of these structures can exist at any instant. Then the molecule resonates between these structures, but this is not superposition?
To me, resonance represents all the possible structures that can simultaneously exist for a single particle, but at any instant the molecule can have only one structure. That is, superposition is different from resonance. But then, again, I am a physical chemist.
Thanks Bryan – resonance structures are a kind of kludge, which have been developed due to the limitations of our system of chemical formulae (and formal charge) in accurately depicting electron densities. The resonance forms are not themselves isolable or “real” but can be thought of as components that contribute to the overall electronic structure of the molecule, which is a weighted hybrid of resonance structures.
I like Bruice”s analogy, here – http://i.imgur.com/tow7V.jpg
You should also include a collection of Common Resonance Errors like trying to draw a pi-bond to pi-bond arrow for an allylic anion, etc.
You have an uncanny ability to read my mind, sir. That is next