Free Radical Reactions
3 Factors That Stabilize Free Radicals
Last updated: May 28th, 2026 |
What Factors Affect Free-Radical Stability?
In the last article we introduced free radicals – neutral, electron-deficient chemical species with a partially filled orbital – and learned that they are highly reactive intermediates in organic chemistry. (See article – Free Radical Reactions)
In this post we’ll cover two of the most important concepts concerning these species: their geometry, and their stability. It’s this latter concept that we’ll see is particularly important for understanding many free-radical reactions in organic chemistry. [Spoiler: the factors that affect free radical stability are largely the same factors that stabilize carbocations [discussed previously here]
Table of Contents
- The Stability of Free Radicals Increases In The Order Methyl < Primary < Secondary < Tertiary
- Free Radicals Are Stabilized By Delocalization (“Resonance”)
- The Geometry Of Free Radicals Is That Of A “Shallow Pyramid” Which Allows For Overlap Of The Half-Filled p-Orbital With Adjacent Pi Bonds
- The Same Factors Which Stabilize Free Radicals Also Stabilize Carbocations
- Notes
- Quiz Yourself!
- (Advanced) References and Further Reading
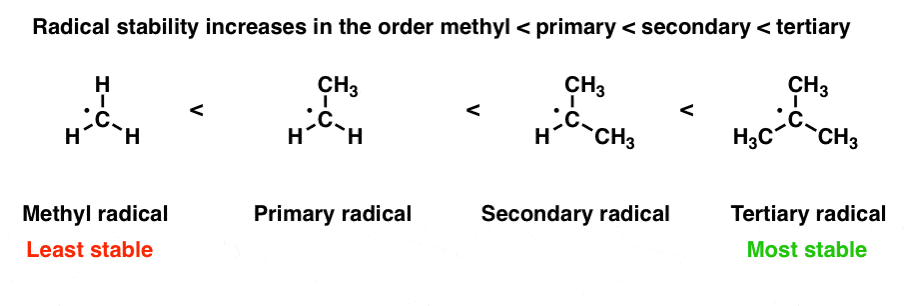
1. Stability Of Free Radicals Increases In The Order Methyl < Primary < Secondary < Tertiary
Let’s talk a bit about stability first, and then circle back to their structure. Being electron deficient, you might already have a hunch regarding factors that might stabilize free radicals. Waaaay back, we talked about how a considerable portion of organic chemistry can be explained simply by understanding that: 1) opposite charges attract (and like charges repel), and 2) the stability of charges increases if it can be spread out over a greater volume. These still apply here!
Electron poor species are stabilized by neighboring atoms that can donate electron density. [“if you’re poor, it helps to have rich neighbors”].
The most common way to interpret “rich neighbors” here is the observation that increasing the number of alkyl groups on the carbon bearing the free radical increases its stability.
Radical stability increases in the order methyl < primary < secondary < tertiary. [For a second, more conceptually complex example, see the bottom of the post]. [Note 1]
2. Free Radicals Are Stabilized by Delocalization (“Resonance”)
Secondly, we have also learned that any factor which can lead to the electron deficient site being delocalized [spread out] over a larger area will also stabilize electron poor species. Previously, for example, we’ve seen that the positive charge of a carbocation is considerably stabilized when it is adjacent to a π bond.
That’s because the carbocation is sp2 hybridized and bears an empty p orbital, allowing for overlap with the adjacent p orbitals and therefore leading the positive charge to be delocalized over multiple carbon atoms, in a manner that is most easily grasped by drawing resonance structures.
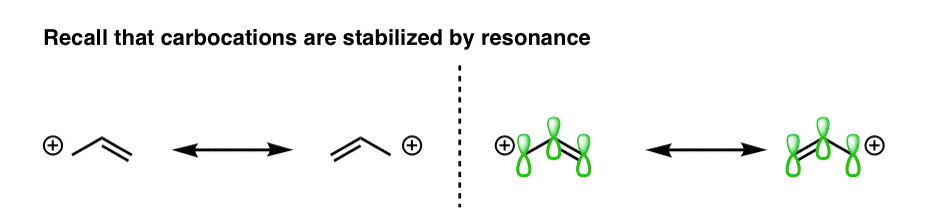
Carbocations are flat – so it’s easy to see how the p orbital could be in line with adjacent p orbitals of a double bond. But what about the geometry of free radicals?
3. The Geometry of Free Radicals Is That Of A “Shallow Pyramid”, Which Allows For Overlap Of The Half-Filled p-Orbital With Adjacent Pi Bonds
If we draw out the electrons in a typical alkyl free radical, we see that there are three bonding pairs and a single unpaired electron, for a total of four occupied orbitals. By analogy to, say, amines, we might expect that the hybridization of the molecule to be sp3 and geometry of a free radical would be trigonal pyramidal.
In fact, the geometry of simple alkyl radicals is very close to flat and is often described as a “shallow pyramid”, with only a slight deviation (~ 5°) from planarity. [Note 2]
When the free radical is adjacent to a π bond, there’s a significant stabilization to be obtained if the p orbitals are all in line so they can overlap [i.e. be “in conjugation”] with each other. Overlap is increased (and the molecule’s energy lowered) if the “shallow pyramid” is flattened out.
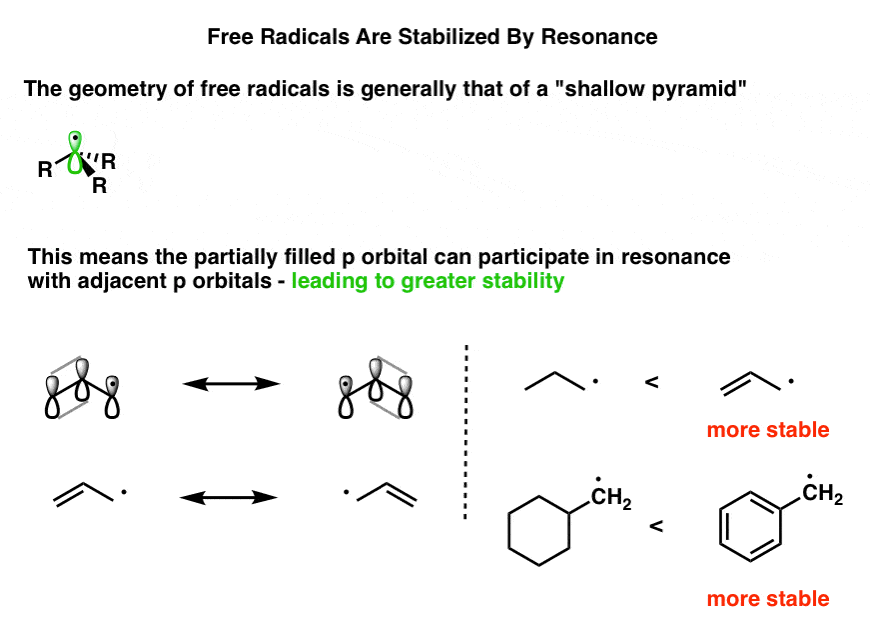
It’s a good approximation to think of a free radical adjacent to a π bond as being sp2 -hybridized.
So what does this all boil down to? The electron-deficient free radical can be delocalized over multiple carbons. Therefore, free radicals are stabilized by resonance.
4. The Same Factors Which Stabilize Free Radicals Also Stabilize Carbocations
If you read the article on the stabilization of carbocations (See article – 3 Factors That Stabilize Carbocations) you might notice something: the same factors which stabilize free radicals are also the same factors which stabilize carbocations!
Quiz time: one of the most stable free radicals known is the triphenylmethyl radical, discovered by Moses Gomberg in 1900. In the absence of oxygen, this radical is indefinitely stable at room temperature. Can you identify the factors which might make this free radical particularly stable?
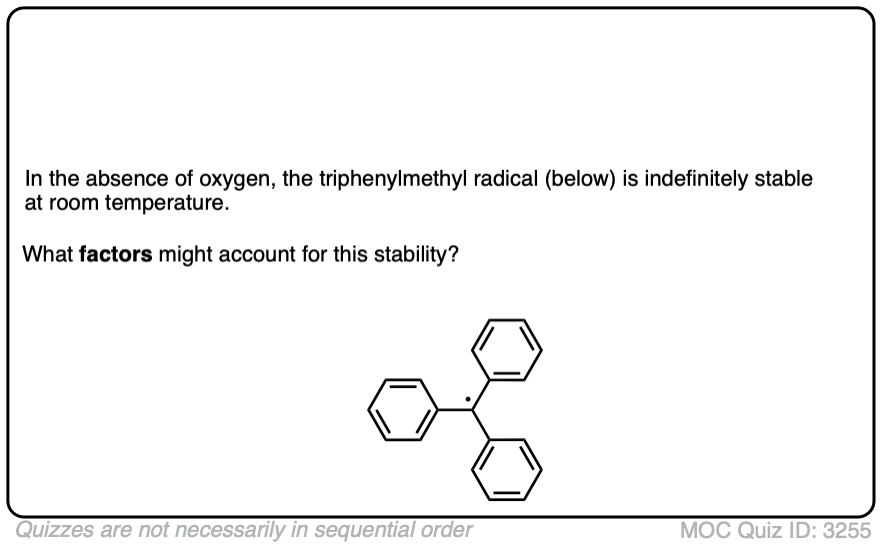 Click to Flip
Click to Flip
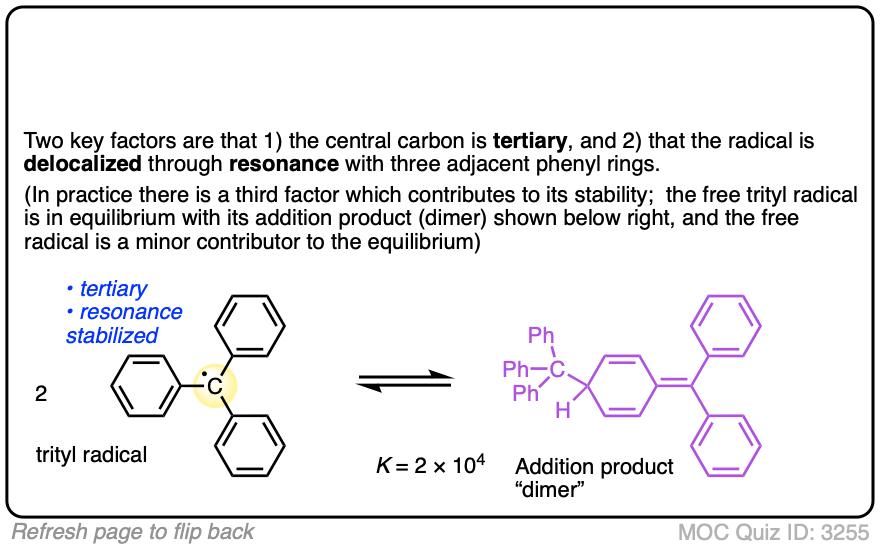
5. Removing Electron Density Destabilizes Free Radicals:
If we remember that free radicals are stabilized by electron donating groups, we might reason that they are destabilized when electron density is taken away.
So what factors might result in a free radical being less “electron rich” ?
There are three major factors. I’ll list them in order of importance for the purposes of a typical student encountering free radicals in a typical class.
6. Radical Stability Decreases With Increasing s-Character Of The Orbital
If you recall some of the factors that affect acidity you might recall that a lone pair of electrons becomes more stable as the hybridization of the carbon goes from sp3 to sp2 to sp. That’s because of the greater s-character of the orbital, which results in the lone pair being held more closely to the (positively charged) nucleus.
What might happen if we’re dealing with a free radical instead? As the s-character of the orbital containing the free radical is increased, the half-filled orbital containing the free radical is held more closely to the nucleus. What effect does this have on the stability of the free radical? It’s actually destabilizing, because being closer to the nucleus, the electron affinity of the orbital will increase.
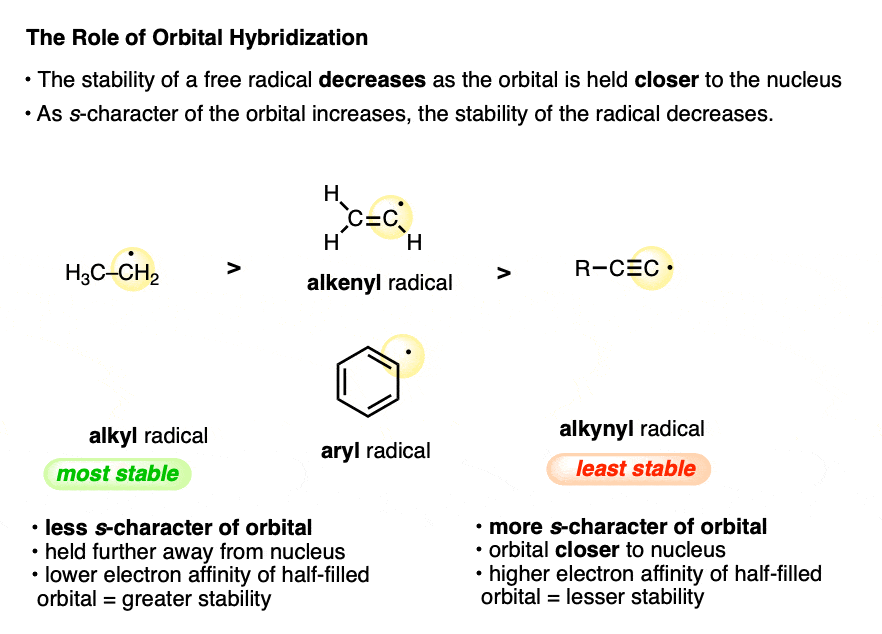
For that reason alkyl radicals (generally considered to be sp2 hybridized) are the most stable, followed by vinyl and phenyl radicals (sp-hybridized) , followed by alkynyl radicals.
7. Radical Stability Decreases With Increasing Electronegativity of The Atom
Quiz time: Having read the paragraph above, what might you think is the effect of electronegativity on free radical stability? What might happen to the stability of a free radical as you increase the electronegativity of the atom? For example, compare the sequence H3C , H2N, HO, and F . Which free radical should be the most stable?

Electronegativity, as we’ve talked about before, is like “greed” for electrons. Increasing electronegativity is going to draw a free radical closer to the nucleus, and as we saw above, this results in destabilization.
8. Radical Stability Decreases As Polarizability Is Decreased
Going down the periodic table, we also notice an increasing stability in free radicals, going from F < Cl < Br < I . While this can likewise be thought of as resulting from a decrease in electronegativity , another way to look at it is that going down the periodic table results in an increase in the size of the atom, and with that, allows for the electron-deficient orbital to be spread out over a greater volume.

9. Summary: Factors Which Stabilize (and Destabilize) Free Radicals
The main factor we’ve seen here that destabilizes free radicals is bringing the half-filled orbital closer to the nucleus (greater s-character, higher electronegativity) or by restricting the delocalization of the free radical (decreasing polarizability). Here, I think it’s important not to focus on the effect of electronegativity on the electron (radical) but on the effect of electronegativity on the “hole” – i.e. the empty orbital. Bringing a half-empty orbital closer to the nucleus will greatly increase its potential energy (the electrostatic attraction of the nucleus for an electron) and increase electron affinity, making that free radical much more reactive (and in this case, reactivity = instability).
So how might we “quantify” the stability of a free radical?
Believe it or not, there’s actually a remarkably simple way to learn how stable free radicals are, using a measurement you’re probably already familiar with! We’ll talk about that in the next post.
Next Post: Bond Strengths And Radical Stability
Notes
Note 1. Stabilization of Free Radicals By Adjacent Lone Pairs
In addition to alkyl groups, free radicals are also stabilized by adjacent groups with lone pairs, such as oxygen and nitrogen. At first thought, oxygen might not seem like much of an electron donating group, since it’s quite electronegative. However, oxygen does have two lone pairs of electrons. How might these be involved?
The adjacent oxygen atom can donate electron density to the half-empty p orbital, which is a stabilizing interaction. The orbital picture looks like this.
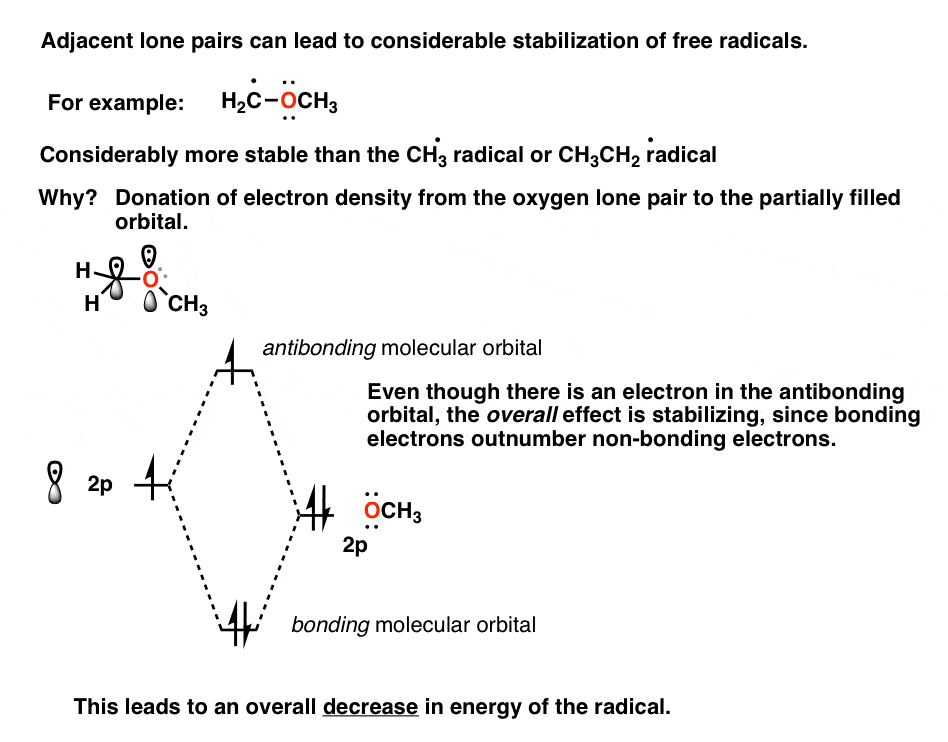
Yes, there’s an electron in the antibonding orbital, but on the whole the interaction is stabilizing since bonding electrons outnumber antibonding electrons here.
Note 2. One note for advanced students – the “shallow pyramid” has a low barrier to inversion (1-2 kcal/mol). This means that if a free radical is formed from an optically active chiral center, rapid racemization generally ensues.
From Carey & Sundberg A, 4th ed. p. 676:
“Simple alkyl radicals are generally pyramidal, although the barrier to inversion is very small…. theoretical results also indicate that the barrier to inversion is no more than 1-2 kcal/mol, so rapid inversion will occur”.
From the same book (p. 675):
“The IR spectrum of the methyl radical has been recorded at very low temperatures in frozen argon. This IR study puts a maximum of ~5° on the deviation from planarity…”
Note 3. A previous version of this post included adjacent electron-withdrawing groups as a destabilizing influence on free-radical stability. I’ve removed this because it’s just too complicated for our purposes.
For example, take the methyl radical, H3C• . Replacing hydrogen with fluorine (a strong electron withdrawing group) one might expect radical stability to decrease. Well, it doesn’t. It’s actually more stable due to the ability of the fluorine lone pair to donate to the half-filled orbital. A second fluorine has a similar effect. However, the F3C• radical is less stable than the methyl radical. This is hard to predict with simple rules. See: Homolytic Bond Dissociation Enthalpies of the C−H Bonds Adjacent to Radical Centers, Xian-Man Zhang The Journal of Organic Chemistry 1998 63 (6), 1872-1877 . DOI: 10.1021/jo971768d
A note on hybridization. Vinyl radicals have an (E) and (Z) form and the inversion barrier from one to the other increases as the electronegativity of the substituents increases. Since the molecule must pass through an sp-hybridized geometry in order to invert, this supports the notion that taking electron density away from the sp-orbital destabilizes the radical. This is a very interesting paper where the rates of inversion were studied:
Effect of Substituents on the Structure of the Vinyl Radical: Calculations and Experiments. Carlo Galli,*, Alessandra Guarnieri,Heinz Koch,†, Paolo Mencarelli,* and, and Zvi Rappoport‡ The Journal of Organic Chemistry 1997 62 (12), 4072-4077 DOI: 10.1021/jo962373h
Quiz Yourself!
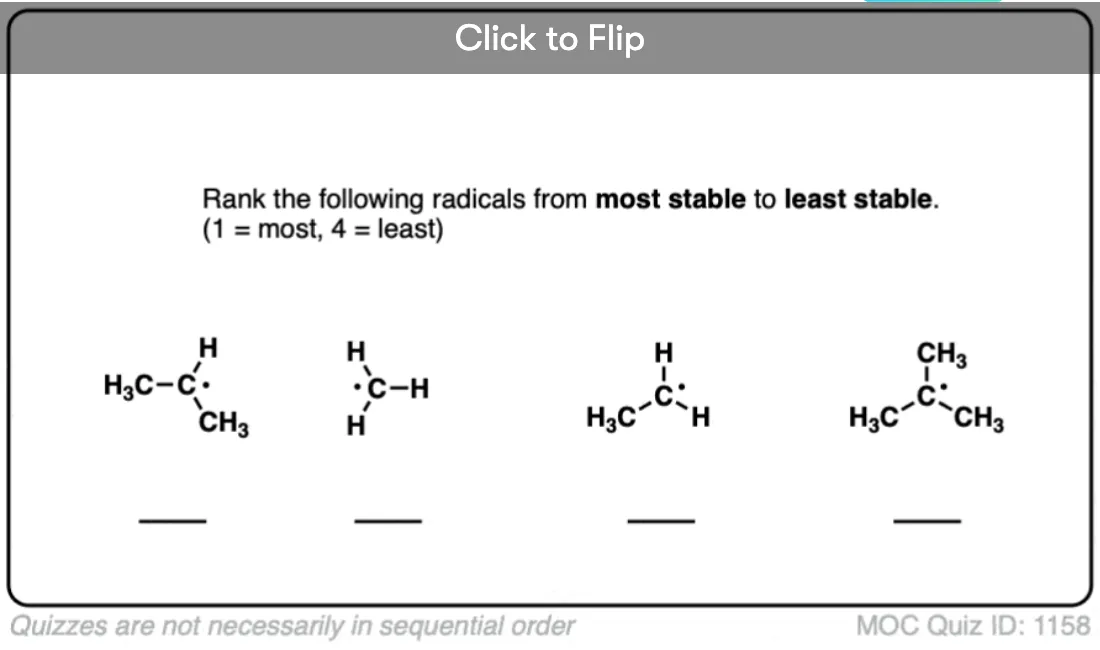
Become a MOC member to see the clickable quiz with answers on the back.
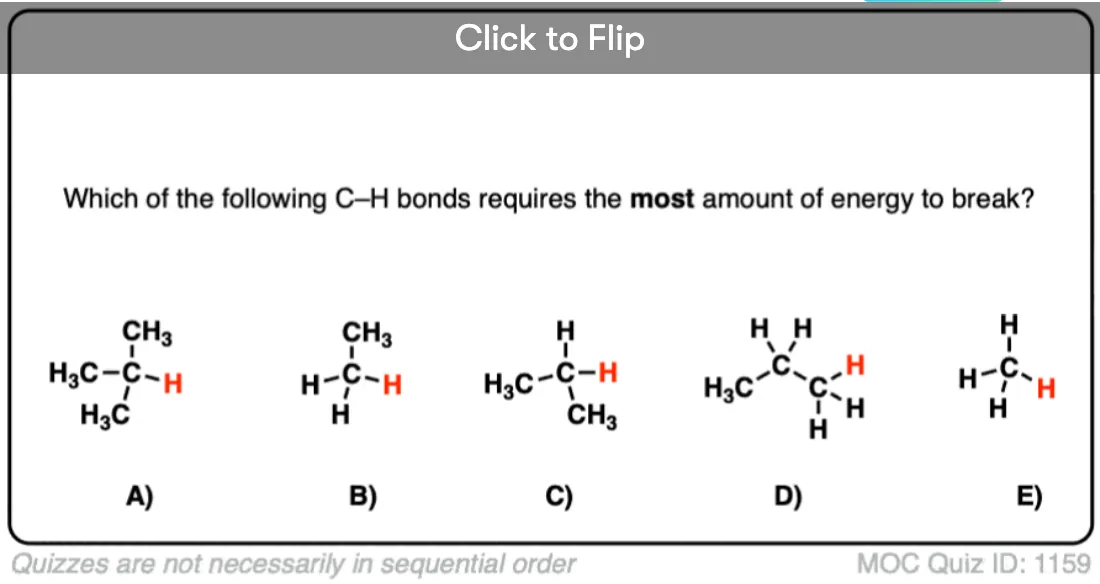
Become a MOC member to see the clickable quiz with answers on the back.
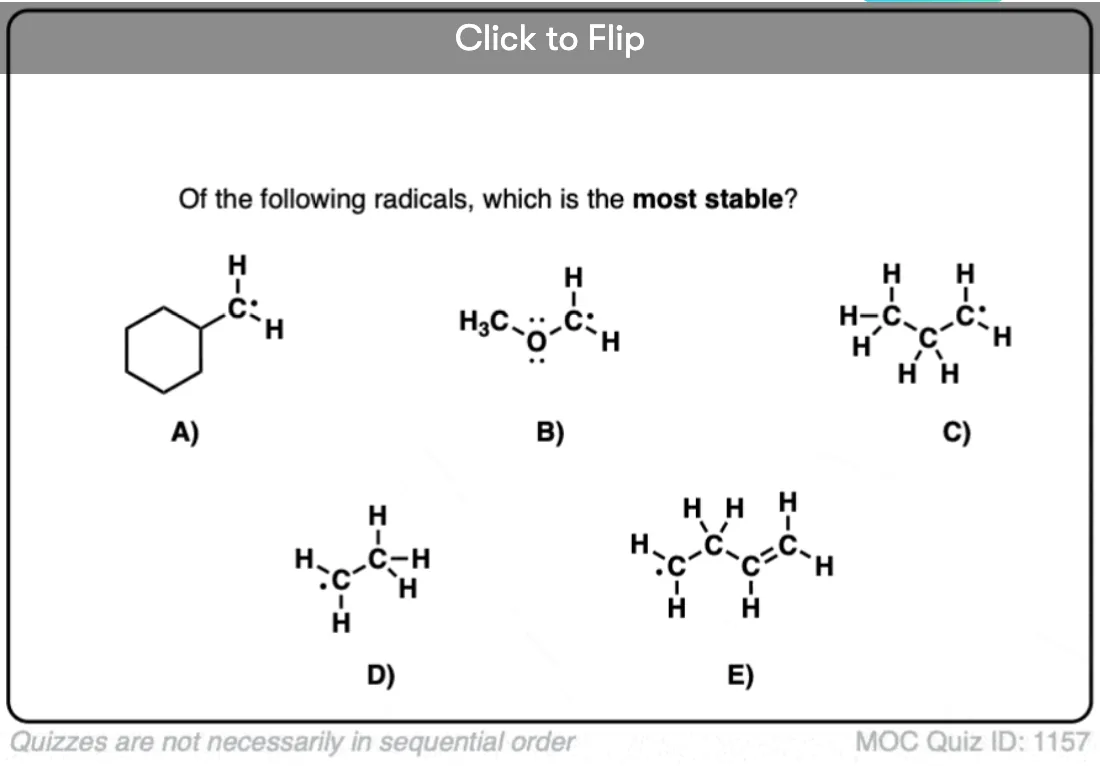
Become a MOC member to see the clickable quiz with answers on the back.
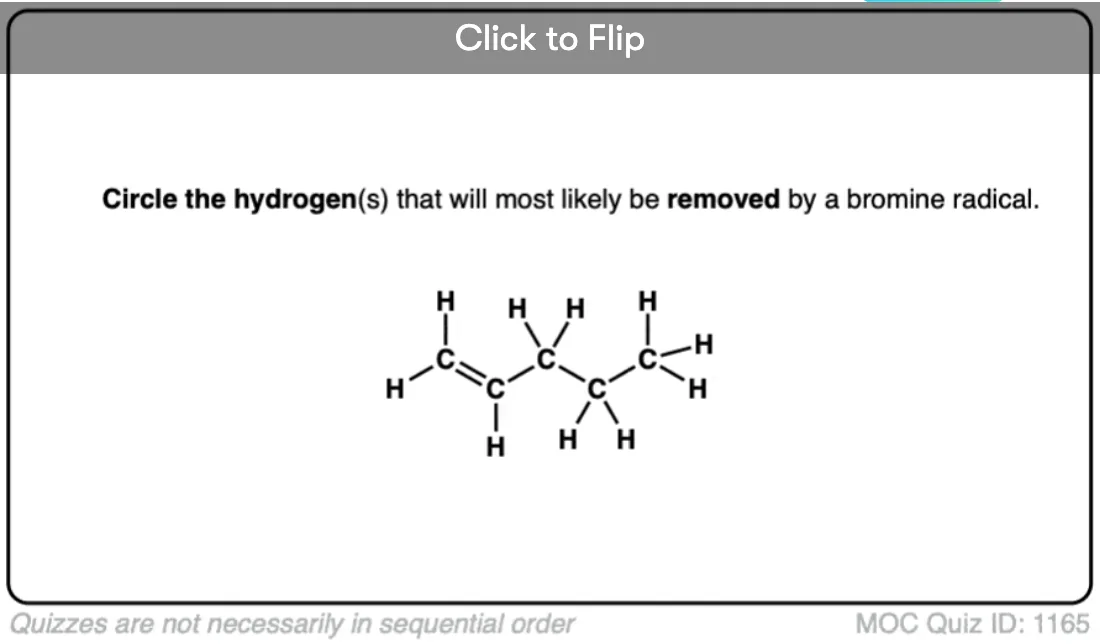
Become a MOC member to see the clickable quiz with answers on the back.
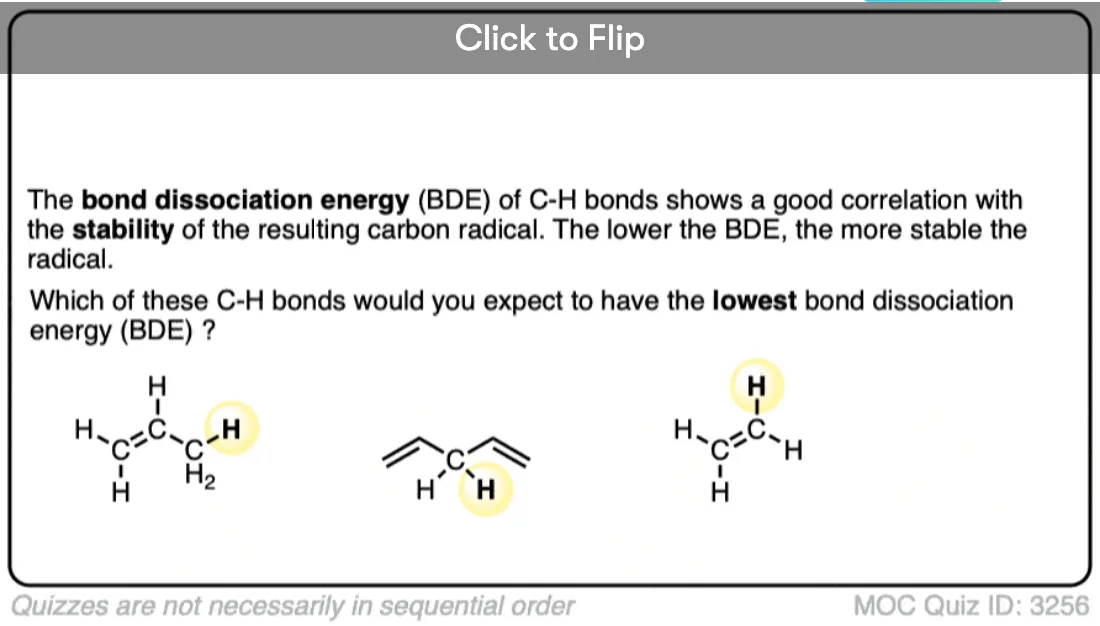
Become a MOC member to see the clickable quiz with answers on the back.
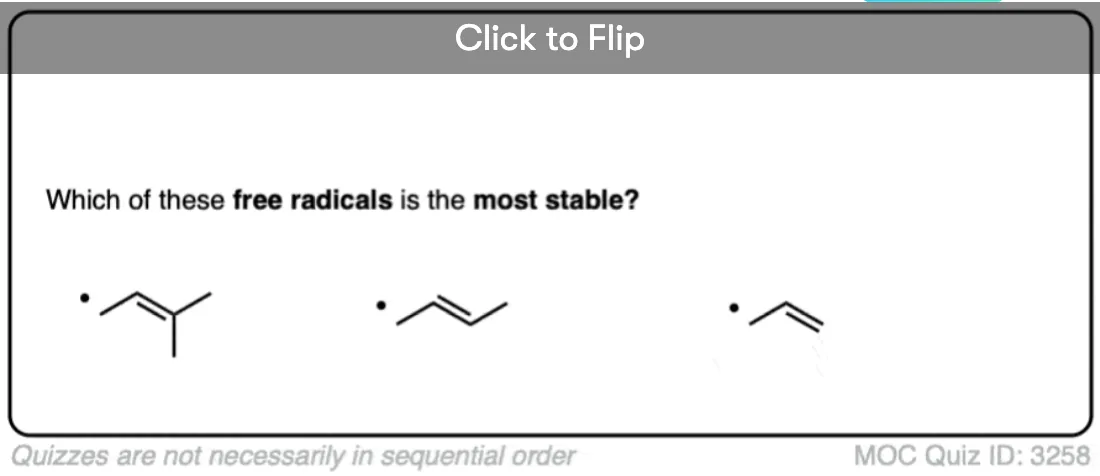
Become a MOC member to see the clickable quiz with answers on the back.
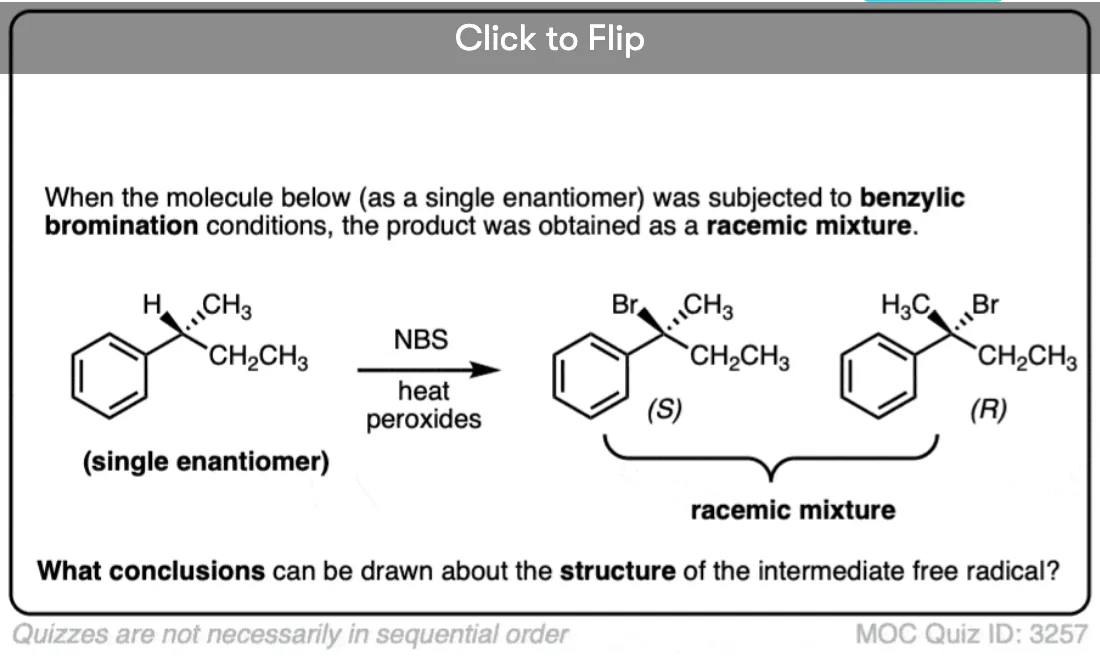
Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading
A useful table of C-H and C-C bond dissociation energies (a useful proxy for radical stabilities) can be found here:
- AN INSTANCE OF TRIVALENT CARBON: TRIPHENYLMETHYL.
Moses Gomberg
Journal of the American Chemical Society 1900, 22 (11), 757-771
DOI: 10.1021/ja02049a006
This is regarded as the famous paper that started the field of organic free-radical chemistry. Moses Gomberg (University of Michigan) was attempting to synthesize hexaphenylethane, and so tried reducing triphenylmethyl chloride (trityl chloride) with Zn metal. Instead, he got a yellow solution which gave a product incorporating oxygen when exposed to air. When he conducted this experiment under a CO2 blanket (remember, pure Ar was not readily available back then!), he found that the solution was indefinitely stable, and deduced that it was the triphenylmethyl radical, which reacted with oxygen when exposed to air, forming a peroxide. At the end of the paper, Gomberg states “This work will be continued and I wish to reserve the field for myself”. The ACS declared the University of Michigan as a national historical chemical landmark in 2000, celebrating the centenary of this discovery, and Prof. Melanie Sanford (U Michigan) currently holds the Moses Gomberg chair in Chemistry. - Theoretical Interpretation of Carbon‐13 Hyperfine Interactions in Electron Spin Resonance Spectra
Martin Karplus and George K. Fraenkel
J. Chem. Phys. 1961, 35, 1312
DOI: 10.1063/1.1732044
The EPR (electron paramagnetic resonance) spectrum of the methyl radical leads to the conclusion that its structure could be either planar or a very shallow pyramid. Prof. Karplus is now at Harvard and received the Nobel Prize in Chemistry in 2013 for his contributions to theoretical chemistry. - Structure of the tert-butyl radical
L. Bonazzola, N. Leray, and J. Roncin
Journal of the American Chemical Society 1977, 99 (25), 8348-8349
DOI: 10.1021/ja00467a052 - Configuration of the tert-butyl radical
D. Griller, K. U. Ingold, P. J. Krusic, and H. Fischer
Journal of the American Chemical Society 1978, 100 (21), 6750-6752
DOI: 10.1021/ja00489a035
The t-butyl radical has been studied extensively. Whereas experimental results have been interpreted in terms of both planar and slightly pyramidal structures, theoretical calculations favor a pyramidal structure. - Reactions of Atoms and Free Radicals in Solution. I. A Study of the Substitution of Hydrogen on an Asymmetric Carbon Atom—The Chlorination of Primary Active Amyl Chloride
Herbert C. Brown, M. S. Kharasch, and T. H. Chao
Journal of the American Chemical Society 1940, 62 (12), 3435-3439
DOI: 1021/ja01869a040 - N-Bromosuccinimide. III. Stereochemical Course of Benzylic Bromination
H. J. Dauben and Layton L. McCoy
Journal of the American Chemical Society 1959, 81 (20), 5404-5409
DOI: 10.1021/ja01529a038
The above two papers demonstrate that radical reactions at chiral carbon centers leads to racemization, because of the easy inversion of the intermediate pyramidal radical. - Ab Initio Calculations of the Relative Resonance Stabilization Energies of Allyl and Benzyl Radicals
David A. Hrovat and Weston Thatcher Borden
The Journal of Physical Chemistry 1994, 98 (41), 10460-10464
DOI: 1021/j100092a014
The stabilization energy of a vinyl group (in the allyl radical) and a phenyl group (in the benzyl radical) has been calculated to be 15.7 kcal/mol and 12.5 kcal/mol, respectively. - Effects of adjacent acceptors and donors on the stabilities of carbon-centered radicals
G. Bordwell, Xianman Zhang, and Mikhail S. Alnajjar
Journal of the American Chemical Society 1992, 114 (20), 7623-7629
DOI: 10.1021/ja00046a003
Table I in this paper contains stabilization energies of methyl radicals with various substituents (e.g. ·CH2X). - Infrared Spectrum of the Methyl Radical in Solid Argon
L. Andrews and G.C. Pimentel,
J. Chem. Phys. 1967, 47, 3637
DOI: 10.1063/1.1712434
This study puts a maximum of about 5° on the deviation of the methyl radical from planarity.
Hi, I have a question: Are charged species with an unpaired electron also free radicals?
There are such things as “radical anions”, which bear both a radical and a charge. One example is the ketyl radical anion that is formed from the reduction of benzophenone by sodium metal. https://en.wikipedia.org/wiki/Ketyl
Is there a free radical activity order for all kinds of radicals? For example,
……> Cl(.) > CH3(.) > Me3C(.) > …..
I don’t understand how do people make remarks on molecular orbitals of heteroatomic species in org. chem. Can somebody explain ?
Why is it that the order of stability of free radicals is allylic > benzylic > tertiary butyl, but is the exact opposite in case of carbocations?
how can factors stabilising radicals and carbocations be same?radicals can be stabilised by both electron donating and withdrawing groups right?
Hi James,
I have several questions regarding the reactivity of the allyl and benzyl radicals towards association with HO2: allyl/benzyl+ho2–>allyl-ho2/benzyl-ho2.
I have read several posts and papers claiming that although the benzyl radical shows more resonant structures than the allyl one, both radicals show similar stability because some of the resonant structures of benzyl are not very stable as they break the aromaticity. This makes me think that the reactivity of both radicals when it comes to associate with an HO2 radical is similar, and both reactions should show similar rate constants.
However, I wonder if this issue could be addressed in a different way. How about looking into the SOMO orbitals of allyl, benzyl, and HO2? I have run quantum calculations and observed that the energy of the SOMO of allyl is 21.9 kcal mol-1 with respect to that of the HO2 (set as 0.0 kcal mol-1 as a reference energy), while that of the benzyl radical is 28.9 kcal mol-1. The energy gap between the SOMO orbitals of the pair HO2-Allyl is 7.0 kcal mol-1 smaller than that of the HO2-Benzyl one. Does this mean that the overlap between the SOMO orbitals of allyl and HO2 is much larger and thus the resulting MO during the association reaction will be much more stable? If so, does this mean that the association reaction allyl+HO2 should show much larger rate constants than benzyl+HO2 reaction?
Thank you
I want to ask when O oxygen shows it +M group character and have free radical on itself by making double bond with tha carbon ,at that time numbers of electronic Oxygen is 9 which can not be possible with the 2nd period’s member….Please explain
Hi, I have questions about the MO diagram: why 4 MO orbitals? and why 6 electrons?
It’s simplified. The interaction is between a lone pair on oxygen and a half-filled orbital on carbon. There are only 3 electrons in the system. The middle section depicts the lowering of the energy of the lone pair, and the raising of the energy of the antibonding orbital. Overall the system is more stable.
Does steric hindrance have any affect in whether a radical will form? For example, would ethylbenzene be more likely to undergo radical halogenation than tert-butylbenzene?
Well, t-butylbenzene would certainly not form a radical as easily as ethylbenzene, but that’s because there are no hydrogens that can be removed that will result in a resonance-stabilized radical!
Generally speaking steric effects are not as important in free radical reactions.
One of the reasons is that charged nucleophiles and electrophiles carry around a solvent “shell” with them that helps to stabilize the charges. Kind of like an entourage that surrounds them at all times. Free radical reactions are neutral and this “entourage” effect is not as much of an issue.
hi..can i ask you one question..why is it when the benzylic undergoes monohalogenation..the halogen attach to the side chain insteaad of the ring itself?
please answer???
Look up the difference between aryl radicals and benzylic radicals. One is resonance stabilized, the other is not.
I want to ask which free radical is the most stable in following
a. Primary
b. Methyl
c. Secondary
d. Tertiary
Plz reply
Can free radicals undergo ring expansion
For our purposes, no.
In fact the cyclopropylmethyl radical is unusually stable, and although I don’t have the numbers at the moment the cyclobutylmethyl radical would also be more stable than an ordinary primary radical as well since it can interact with the “banana bonds” of the cyclobutane.
If I want to irradiate UV to polypropylene (PP, it has tertiary C) and polysulfone (PSf, it has quarternary C), would PSf form free-radical more easily?
Thanks for taking time to answer my question.
Yes PSf certainly would form a radical more easily since it has a chromophore that can be accessed with UV light. You’d get loss of SO2 and formation of two adjacent radicals which could recombine or go on to other chain reactions, depending on conditions.
Could you please explain why are there two unpaired electrons in the MO diagram at the end of your article? Shouldn’t it be only one?
Hi – this is an effect called “hyperconjugation”. It’s an interaction between a filled orbital (the lone pair of oxygen) and a half-empty orbital (the free radical). Donation of electron density to the half-filled orbital results in some stabilization. This is what the orbital picture is trying to present. I haven’t written an article on hyperconjgation yet but there is a wikipedia article.
Thanks for clarifying.
MO diagrams with unpaired electrons and molecules with more than two atoms are unfamiliar to me, so I’ll settle for that for now :)
This “hyperconjugation” that James was talking about is also the reason for the stability increase from primary to tertiary alkyl radicals; it’s worth noting that the lone pair on the oxygen and the two electrons in the sigma bond between the adjacent carbon and hydrogen are carrying out the same function, and aren’t separate entities.
I wouldn’t call it hyperconjugation since hyperconjugation is (by definition) between sigma bond and and empty or half empty orbital. A quantum phyical chemist would call it just conjugation. Even though it’s very unorthodox.
[Modern Physical Organic Chemistry By Eric Anslyn]