Organic Chemistry Tips and Tricks
Alkene Addition Pattern #2: The “Three-Membered Ring” Pathway
Last updated: January 23rd, 2024 |
The “Three-Membered Ring” Pathway In Alkene Mechanisms: Halogenation, Oxymercuration, Halohydrin Formation, and Acidic Epoxide Opening
In the last post we walked through a proposal for how the bromination of alkenes works and showed that it adequately explains many of the experimental observations made for this reaction.
Namely, the reaction proceeds with anti addition of substituents across the alkene, and (where relevant) the reaction proceeds with “Markovnikov” regioselectivity. These observations are best explained through the intermediacy of a “bromonium ion”.
In this post we try to show that bromination is but one example of a whole family of reactions in introductory organic chemistry that pass through a positively charged three membered ring, including not just halogenation, but oxymercuration, halohydrin and haloether formation, and even addition to protonated epoxides.
Since all of these reactions follow the same pattern, if you learn any one of these mechanisms, you’ve essentially learned them all!
Table of Contents
- Bromination of Alkenes: The Mechanism
- Chlorination of Alkenes: Mechanism
- Iodination of Alkenes: Mechanism
- Chlorohydrin Formation: Mechanism
- Haloether Formation: Mechanism
- Chlorohydrin Formation With NBS: Mechanism
- Oxymercuration Of Alkenes: Mechanism
- Oxymercuration: The Reduction Step
- A Non-Obvious Cousin Of Halonium Ions: Protonated Epoxides
- Summary: The Key Pattern Of The “Three-Membered Ring Pathway”
- Notes
- (Advanced) References and Further Reading
1. Bromination of Alkenes: The Mechanism
This is just a review of what we saw in the previous post. Treating an alkene with Br2 results in a vicinal dibromide with the two bromines oriented anti to each other. The key intermediate is a “bromonium ion”, which contains a positively charged 3-membered ring.
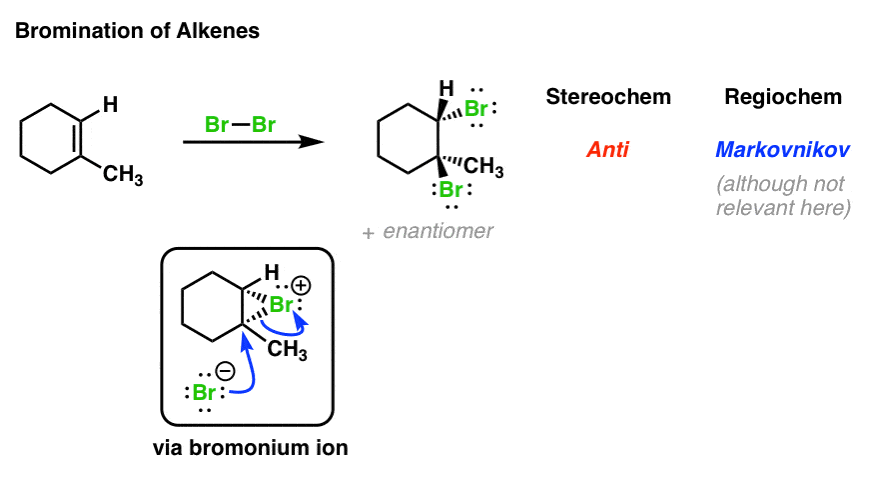
Taking bromination of alkenes as a starting point, we might ask: “do variants of this mechanism operate for other reactions of alkenes as well?”
The answer is yes!
2. Chlorination of Alkenes: The Mechanism
Take, for example, the chlorination of alkenes. The products of this reaction has identical patterns of stereoselectivity and regioselectivity to those of bromination. Therefore we might surmise that they proceed through the same type of reaction intermediate! [This intermediate is called the “chloronium ion”]
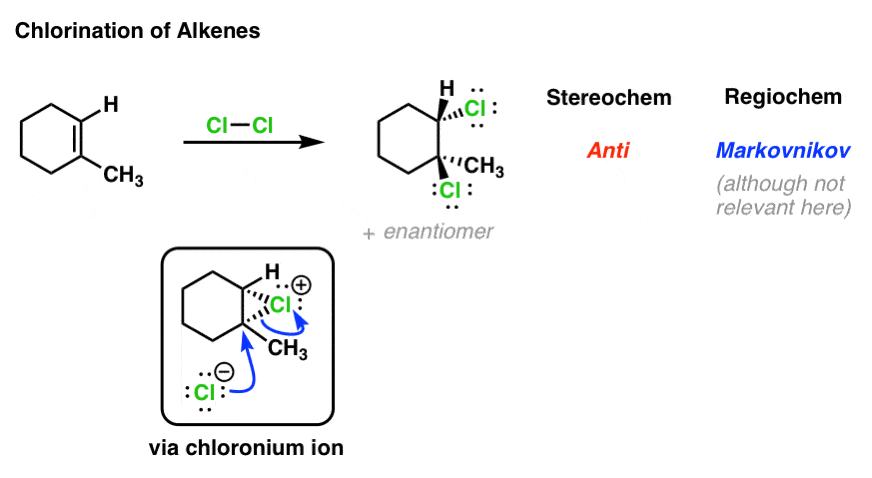
3. Iodination of Alkenes: The Mechanism
This is also the case for iodination reactions, which proceed through the “iodonium ion”:
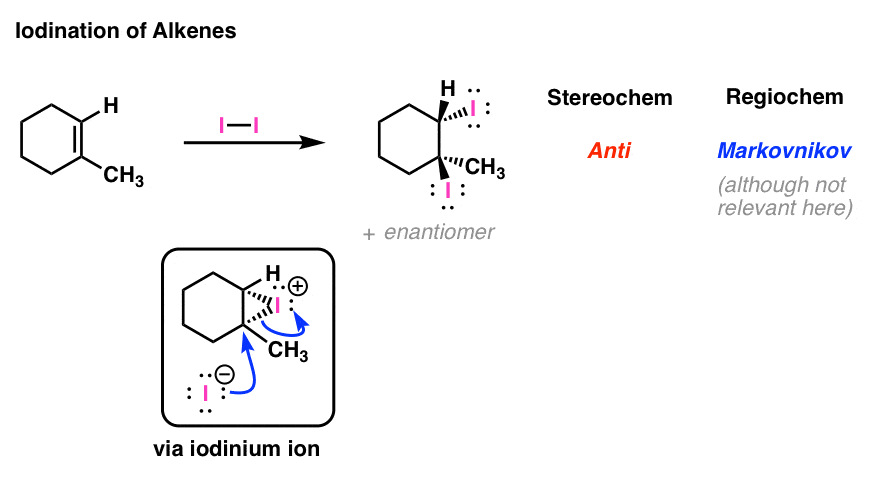
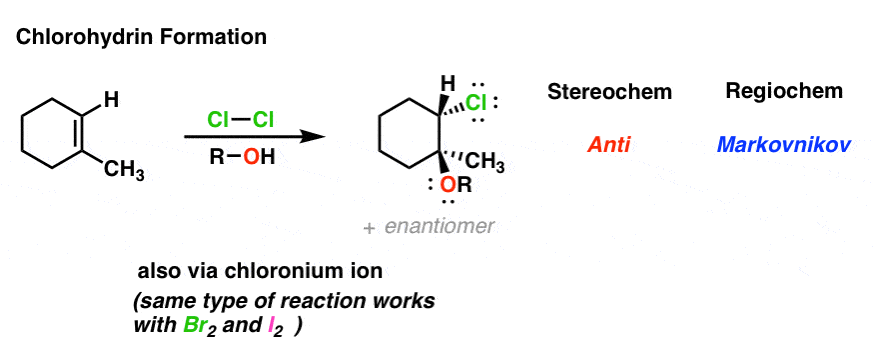
4. Chlorohydrin Formation: The Mechanism
This also applies to reactions where the intermediate chloronium ion is trapped with solvent (water in this case). After deprotonation of R–OH+ to give R–OH, the product is referred to as a “chlorohydrin”.
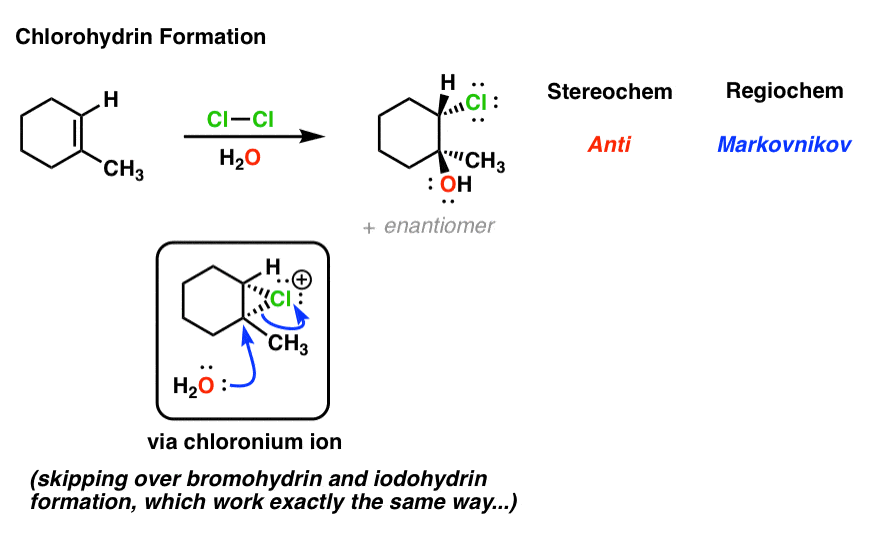
Note that bromohydrin and iodohydrin formation work exactly the same way and if you merely replace “Cl” with either of those halogen atoms you’ll obtain the indicated product.
5. “Haloether” Formation: The Mechanism
As described in the last few posts, what’s notable about bromination is that by using a solvent which can act as a nucleophile, we can obtain products which incorporate that solvent. For example by using an alcohol as solvent, we obtain the following “chloroetherification” product. It likewise proceeds through the exact same mechanism described above.
Furthermore, these reaction pathways are not confined to the dihalogens Cl2, Br2, and I2 [nor F2, the Tiger of Chemistry, which is a very difficult beast to keep on its leash]. And a good thing too, since Cl2 and Br2 are to various extents vile and inconvenient to work with.
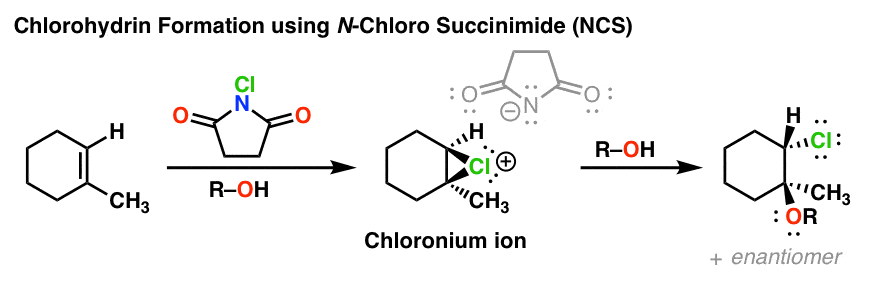
6. Chlorohydrin Formation Using N-Chloro Succinimide
A convenient source of “electrophilic” chlorine is the crystalline salt N-chlorosuccinimide (NCS), an innocuous appearing white crystalline solid. Alkenes react rapidly with NCS to form chloronium ions, which can then be intercepted to form a variety of useful products by analogy to those shown above. With the exception of this more convenient source of halogen, the reaction is otherwise the same. N-bromosuccinimide (NBS) and N-iodosuccinimide (NIS) likewise find use.
Moving beyond the halogens, are there other reagents that form these cyclic intermediates? Why, yes indeed.
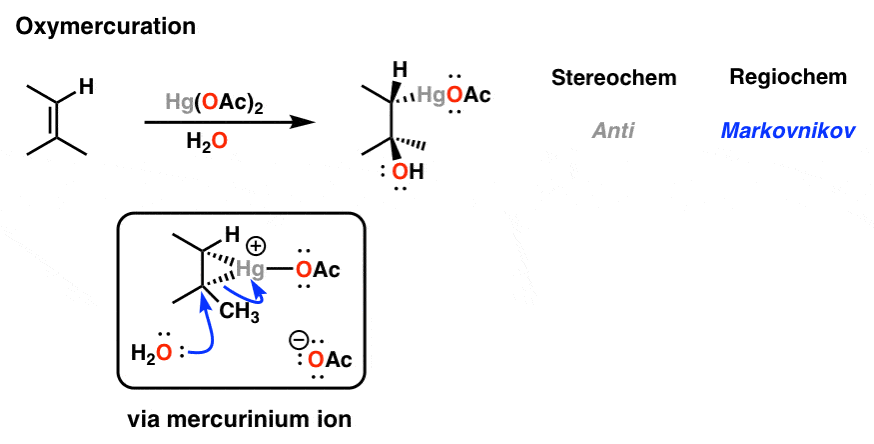
7. Oxymercuration: The Mechanism
When alkenes are treated with mercury (II) salts (such as mercuric acetate) in the presence of water or alcohols, we obtain products with the same pattern of stereochemistry and regiochemistry that we’re accustomed to seeing by now. What’s a likely intermediate here? A three-membered ring called the “mercurinium ion”.
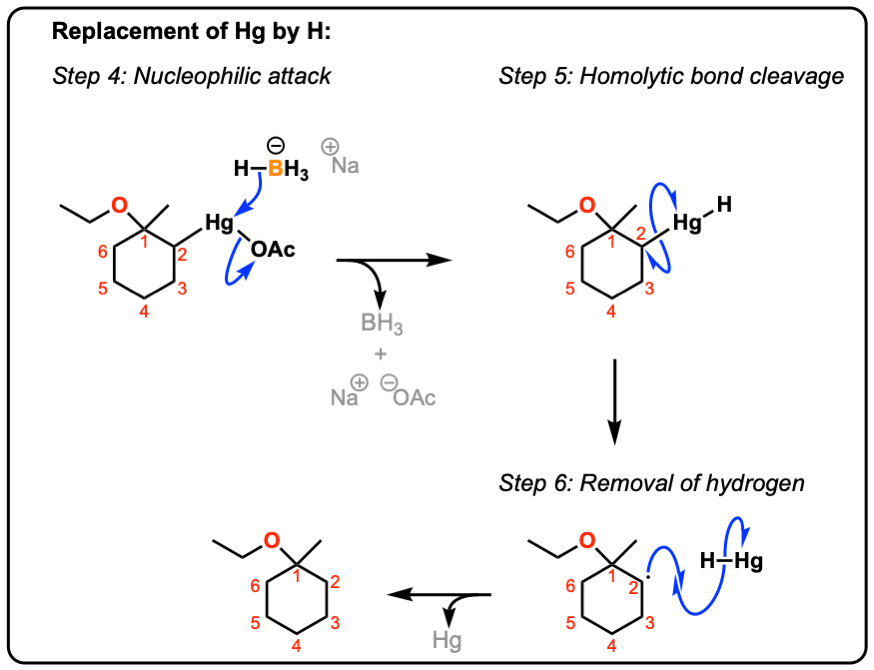
8. Oxymercuration: The Reduction Step
Organomercury compounds find very little application in themselves, but can be used as intermediates in subsequent reactions. To replace mercury with hydrogen, sodium borohydride (NaBH4) is added. In this case, rather than being “anti” , the stereochemistry of this reaction ends up being a wash: treatment with NaBH4 leads to cleavage of the C-Hg bond and formation of a free radical. The free radical can react from either face with hydrogen, leading to scrambling of the stereocenter.
To see a plausible mechanism, hover here or click this link.
{kind=link}
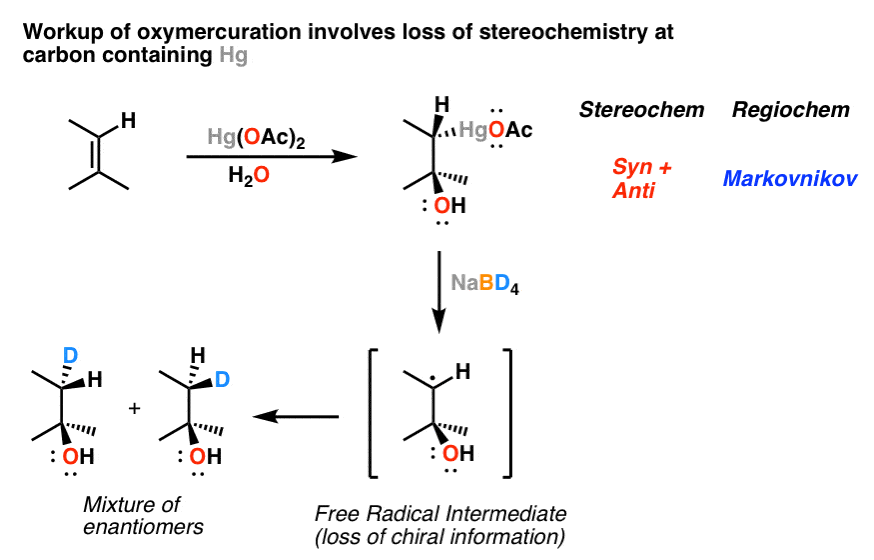
How many other reactions go through this type of mechanism? There is actually a sizable list. For instance, there are electrophilic sources of sulfur and selenium that can likewise form three membered cationic rings just like those we’ve seen; we won’t go into those.
9. A Non-Obvious Cousin: Protonated Epoxides
One last example that is worth going into is one that might not immediately seem obvious: protonated epoxides.
Treatment of an epoxide with acid leads to a positively charged intermediate that resembles a bromonium ion. As you might guess, the nucleophile attacks the backside of the most substituted carbon and the resulting product has anti stereochemistry. Just as we’ve seen numerous times above.
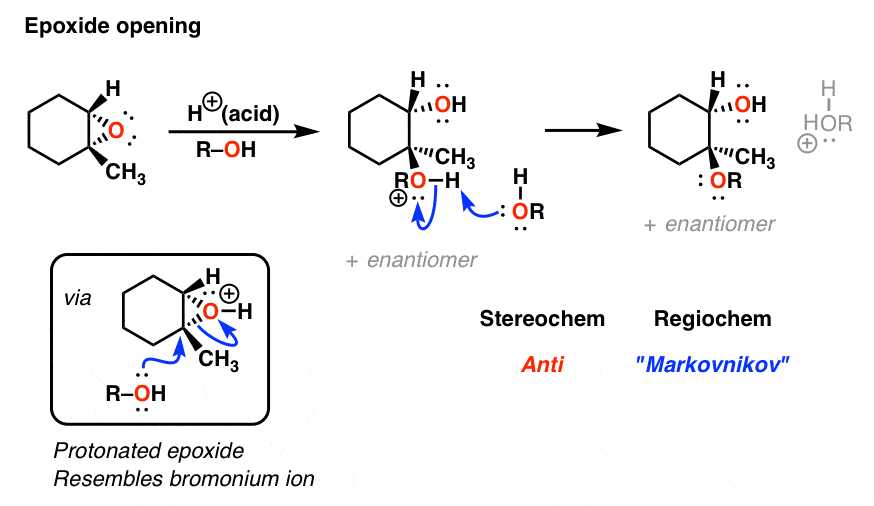
10. Summary: The Key Pattern Of The “Three-Membered Ring Pathway”
Do some of the images in this post look repetitive? They should!
The lesson for this very long post from today is that one can group together a sizable number of different reactions by identifying their common mechanism. Just as there is a family of reactions that pass through the carbocation pathway, there is likewise a “family” of reactions that pass through a three-membered ring. Instead of learning a dozen different mechanisms, we merely learn one – and merely change the actors to suit the occasion.
NEXT POST – Hydroboration of Alkenes
Notes
Note 1. Besides the dihalides, there are also such things as mixed dihalides, such as iodine monochloride. We have all the tools at our disposal to answer how this reaction might proceed. What do you think the product is?
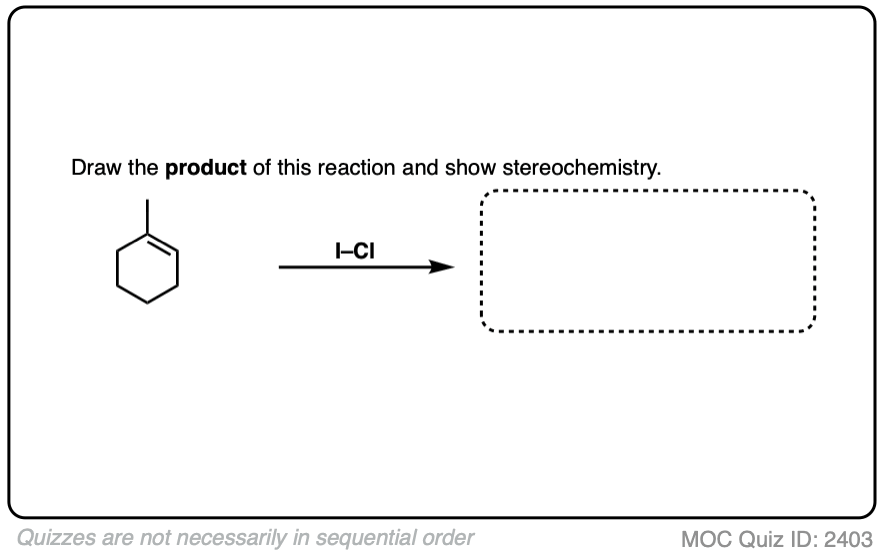 Click to Flip
Click to Flip
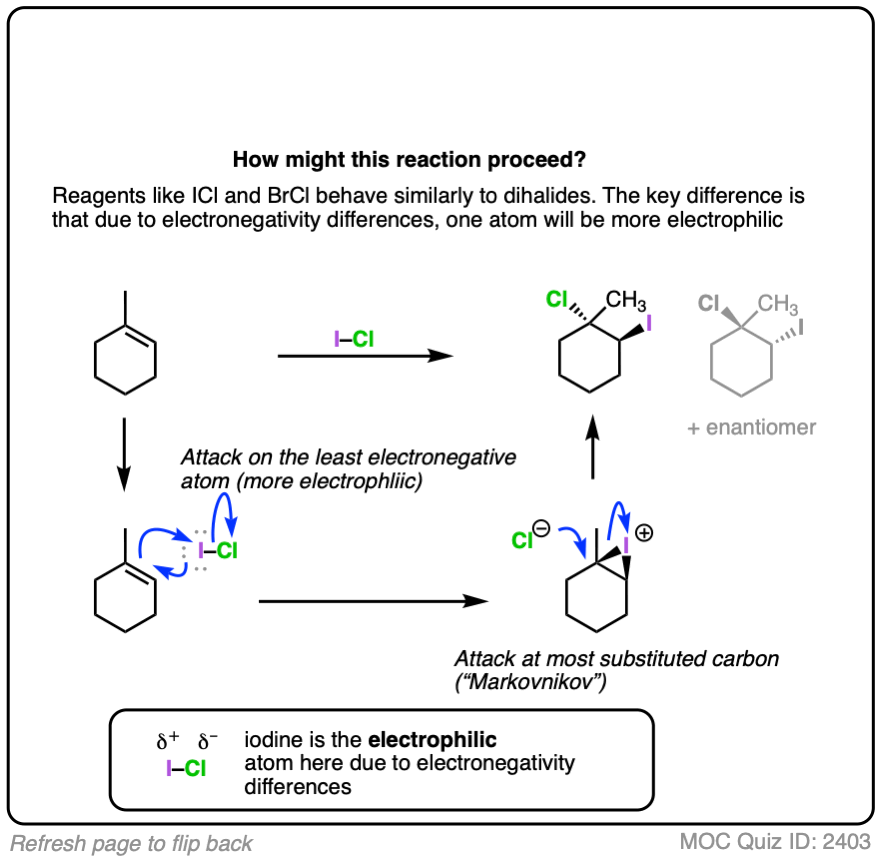
(Advanced) References and Further Reading
This is a common mechanism for several reactions, including halogenation, halohydrin formation, and oxymercuration.
Halogenation:
- The Halogenation of Ethylenes
Irving Roberts and George E. Kimball
Journal of the American Chemical Society 1937, 59 (5), 947-948
DOI: 1021/ja01284a507
One of the earliest descriptions in the literature of a three-membered bromonium ion, accounting for the anti stereochemistry of this reaction. - Stable carbonium ions. LXII. Halonium ion formation via neighboring halogen participation: ethylenehalonium, propylenehalonium, and 1,2-dimethylethylenehalonium ions
George A. Olah, J. Martin Bollinger, and Jean Brinich
Journal of the American Chemical Society 1968, 90 (10), 2587-2594
DOI: 1021/ja01012a025
This is an early paper on the characterization by NMR of halonium (3-membered chloronium, iodonium, and bromonium) ions by ionization of 1,2-dihaloethanes in superacid medium (SbF5/SO2). - Investigation of the Early Steps in Electrophilic Bromination through the Study of the Reaction with Sterically Encumbered Olefins
R. S. Brown
Accounts of Chemical Research 1997, 30 (3), 131-137
DOI: 10.1021/ar960088e
An account by R. S. Brown describing the research he carried out in interrogating bromonium ion intermediates by a variety of methods.Mercuration: - Mechanism of the oxymercuration of substituted cyclohexenes
Daniel J. Pasto and John A. Gontarz
Journal of the American Chemical Society 1971, 93 (25), 6902-6908
DOI: 1021/ja00754a035
This paper demonstrates that “the oxymercuration of substituted cyclohexenes proceeds via mercurinium ion intermediates which are formed in fast, reversible pre-rate-determining step equilibria”. - Organometallic chemistry. IV. Stable mercurinium ions
George A. Olah and Paul R. Clifford
Journal of the American Chemical Society 1973, 95 (18), 6067-6072
DOI: 1021/ja00799a038
The above paper by Nobel Laureate Prof. G. A. Olah demonstrates the existence and intermediacy of mercurinium ions in these reactions via the s and p routes using NMR spectroscopy. - Solvomercuration-demercuration. 11. Alkoxymercuration-demercuration of representative alkenes in alcohol solvents with the mercuric salts acetate, trifluoroacetate, nitrate, and methanesulfonate
Herbert C. Brown, Joseph T. Kurek, Min Hon Rei, and Kerry L. Thompson
The Journal of Organic Chemistry 1984, 49 (14), 2551-2557
DOI: 1021/jo00188a007
While Hg(OAc)2 is the most commonly used reagent for this purpose, the trifluoroacetate, trifluoromethanesulfonate (triflate), or nitrate salts are more reactive and may be preferable for certain applications. - Mechanism of reduction of alkylmercuric halides by metal hydrides
George M. Whitesides and Joseph San Filippo Jr.
Journal of the American Chemical Society 1970, 92 (22), 6611-6624
DOI: 1021/ja00725a039
The reduction (demercuration) step is complex and involves free radicals. This paper by Prof. Whitesides (MIT, now at Harvard) studies the mechanism of this reduction, which is mentioned inside.
Really great work sir
Hi im having a hard time with Br2 (or any halogenation reaction) followed by an INTRAmolecular nucleophilic reaction. Such as Br2 added to a alkene that has a carboxylic acid attached? i know it forms a cyclic product but will it be a an ester type product?
Yes it will! It will form a cyclic ester known as a “lactone”. This reaction is specifically called “halolactonization”. Iodolactonization is a prominent example. https://en.wikipedia.org/wiki/Iodolactonization
… unless because it is in acid… maybe the alcohol on the more substituted carbon then gets protonated again (it is in acid) and leaves forming a carbocation, so the epoxide reforms and is now scrambled in stereochemistry (because it can attack the carbocation from below or above the ring) and the reaction with alcohol then occurs again so you get enantiomers… I dunno…
Karin,
I’m with you — I don’t think you get enantiomers with epoxide + acid… also I think James agrees (unless I’m missing something) in his post here
https://www.masterorganicchemistry.com/2015/02/02/opening-of-epoxides-with-acid/
Thank you for your post; it was very helpful!
I’m just a bit confused on how to draw anti vs. syn in an opened chain. Why would it differ if it was anti or syn if the bond is in free rotation? how can we depict it in the drawing?
Again, thank you!
In the last reaction, why would the final product have Cl bonded with the tertiary carbon and not with either the tertiary or the secondary one?
A minor thing that keeps appearing unclear to me is the projections of some of the atoms. In your final products of oxymercuration the dash on the methyl group is drawn as a straight line now. Is this me overthinking your drawing or is there something I’m legitimately missing? I’ve been seeing minor changes like that in lots of instances (not just on here) and it’s slightly confusing me since I’m interpreting the C as sp3 which would make it tetrahedral, correct? So should still be a dash while the OH group remains a wedge as it is currently drawn. Thanks!
It’s a valid question. I’d start by asking, “what’s the purpose of dashes and wedges” ? And I’d say that ultimately it’s to resolve ambiguity. If there’s more than one way to arrange the groups on a carbon, then dashes and wedges are required to unambiguously depict which configuration we’re dealing with.
On the example you mention, the carbon is attached to two methyl groups and an OH, and there’s only one way to draw that configuration. It would not be incorrect to continue to draw that CH3 as a dash, but it would be more work, and since it’s not necessary, I just didn’t do it.
The carbon is tetrahedral, yes, but that’s always assumed for an sp3 carbon.
I think your NCS thingie is incorrect . NCS gives chlorine free radical and not chlorinium ion
I disagree. That would give a nitrogen-centered radical, which in turn is not stabilized by resonance. NCS in the presence of acid can provide a source of Cl2, which itself can dissociate and be a source of Cl free radical.
Why does the nucleophile attack the ring at the MORE substituted carbon?
Can you post the answer to the post question where iodine monochloride reacts with 1methyl cyclohex2ene?
The double bond attacks iodine, and chlorine is the leaving group. [it attacks iodine because iodine is more electron poor due to the more electronegative chlorine taking electron density away]. Then Cl- attacks the most substituted carbon from behind. Answer here http://imgur.com/rKzsQPo
With ICl, you actually get close to a 50:50 mixture on 1-methylcyclohexene derivatives. With BrCl, you get good selectivity for the bromine at the more substituted carbon, which suggests that the chloronium forms, then bromide acts as the nucleophile. This is explained by the fact that the Br- is a more stable leaving group, even though it is less electronegative.
Very interesting, did not know that. Thank you.
I don’t really understand how two enantiomers can be formed during an epoxide opening. Since the epoxide is already the three-membered ring, I would expect that attack of the alcohol is only possible from underneath the epoxide ring. If it would attack from the same angel as the epoxide is than the reaction is not “anti” anymore. I would therefor expect that only 1 structure can be formed from a specific epoxide.
Or does it mean that the alcohol can attack both at the tertiary carbon and the secondary carbon. I wouldn’t expect this because this would be against the Markovnikov rule, is it not?
How would the mixed dihalide reaction proceed?
Also through a 3 membered intermediate, and also with anti stereochemistry. Key is to figure out which of the halogen atoms is the electrophile, which you can do by comparing their relative electronegativities.
why is markovnikov relevant in a few reactions above and not in others ,markovnikov is relevant when th ydrigen bonds to the most carbon with most hydrogen’s but here markovnikov is relevant even when hydrogen’s are not being bonded
It’s not relevant when we’re adding the same group to both sides of the alkene (e.g. Br2)
Would you consider writing a post about the demercuration mechanism? It’s one of the mechanisms that has bugged me most since it was not taught in any of my organic classes.
Or, if not, do you know of any papers or other sources that show it?
Thanks a lot!
Included a link – thanks for mentioning this, it was definitely something I wondered about when learning this reaction.
I think a radical on the Hg-H is missing in the link with the mechanism of the C-Hg bond reduction.
Good point – but the drawing is correct! The drawing shows that we go from Hg in the (II) oxidation state to Hg in the (I) oxidation state. Similarly, at the end of the complete process we have Hg (0) . We *could* draw in those electrons, but generally do not. For example, when we draw FeCl3 we don’t draw in all the electrons on the Fe atom, we can figure that out by knowing where Fe is on the periodic table and that it is in the (III) oxidation state.
Is there evidence of radical and/or Hg(I) intermediates in the demercuration step? I would think C-H bond forming reductive elimination from Hg(II) would be a possibility after making the mercury hydride. Do you have a link to the primary lit. that shows the scrambling of stereochem with the isotopic label?
Yes, there’s lots of evidence that demercuration is a free radical process.
Whitesides: J Am Chem Soc 1974, vol 96 p 870
evidence – can be diverted by oxygen; also, cyclic products were observed when hex-5-enylmercury compounds were reduced with sodium borohydride [ this doesn’t occur in the presence of oxygen, indicating that trapping is much faster than cyclization]
JACS 1976 vol 98 p. 5973
It’s possible to demercurate w/o scrambling by using sodium amalgam.
My source for this is Carey & Sundberg B 4th ed.