Reactions of Aromatic Molecules
More Reactions on the Aromatic Sidechain: Reduction of Nitro Groups and the Baeyer Villiger
Last updated: May 28th, 2026 |
Here we cover three new reactions of aromatic substituents:
- reduction of nitro groups to amines,
- protection of amines as amides,
- and the Baeyer-Villiger oxidation of ketones to esters
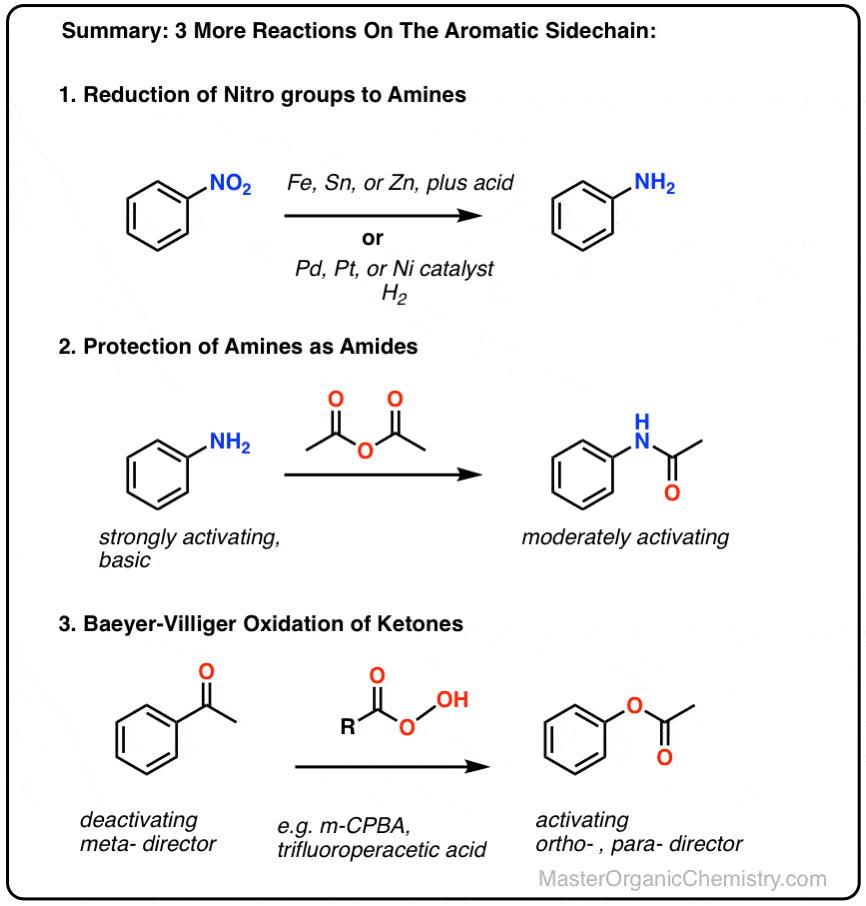
Table of Contents
- Three More Reactions of Aromatic Substituents
- Reduction of Nitro Groups
- Protection of the Amino Group
- The Baeyer-Villiger Reaction
- Mechanism of the Baeyer-Villiger Reaction
- Migratory Aptitudes In The Baeyer-Villiger
- A Note of Caution About Mixing Up “Amides” and “Esters”
- Summary: Reduction of the Nitro Group and The Baeyer-Villiger Reaction
- Notes
- Quiz Yourself!
- (Advanced) References and Further Reading
1. Three More Reactions Of Aromatic Substituents
In this section on reactions of aromatic compounds, we’ve seen two important classes of reactions:
- First, there’s aromatic substitution reactions, where bonds form and break directly on the aromatic ring, resulting in new substituents. We see this in Electrophilic Aromatic Substitution, for example [See post: Introduction to Electrophilic Aromatic Substitution]
- Second, there’s all the reactions of the substituents on aromatic rings. We covered reduction of carbonyls to methylene (CH2) carbons in a previous post [link], as well as oxidation of benzylic C-H bonds to C-Br and C-O bonds [link].
Reactions of the substituents is an important section to master, because modifying the substituents can dramatically change how the aromatic ring undergoes electrophilic aromatic substitution reactions.[see: Understanding Ortho, Para, and Meta Directors]
For instance, we saw that reducing the C=O to CH2 has a tremendous impact on the reactivity of the ring, since it converts a deactivating meta– director into an activating ortho- , para– director.
In this post, we’ll conclude this sub-topic with three more reactions of aromatic substituents, including:
- reduction of nitro groups to amines,
- protection of amines,
- and conversion of ketones to esters, a reaction known as the Baeyer-Villiger Oxidation.
Why are these reactions important?
The ultimate aim of learning reactions is to able to apply them in the synthesis of molecules of our own design from simpler, cheaper components. Each reaction is a “tool” that serves a specific purpose in allowing the formation and breakage of a certain characteristic set of bonds. The more tools you have in your “shop”, the greater the power you have in designing syntheses of organic compounds.
2. Reduction of Nitro Groups
One of the most important reactions of aromatic substituents is the reduction of nitro groups to amines. This can be done using two general methods:
- Addition of an easily oxidized metal like iron (Fe), tin (Sn) or zinc (Zn) in the presence of acid, such as HCl (but often just written, “H+ “) will convert NO2 to NH2.
- Hydrogenation over a palladium, platinum, or nickel catalyst will also convert NO2 to NH2.
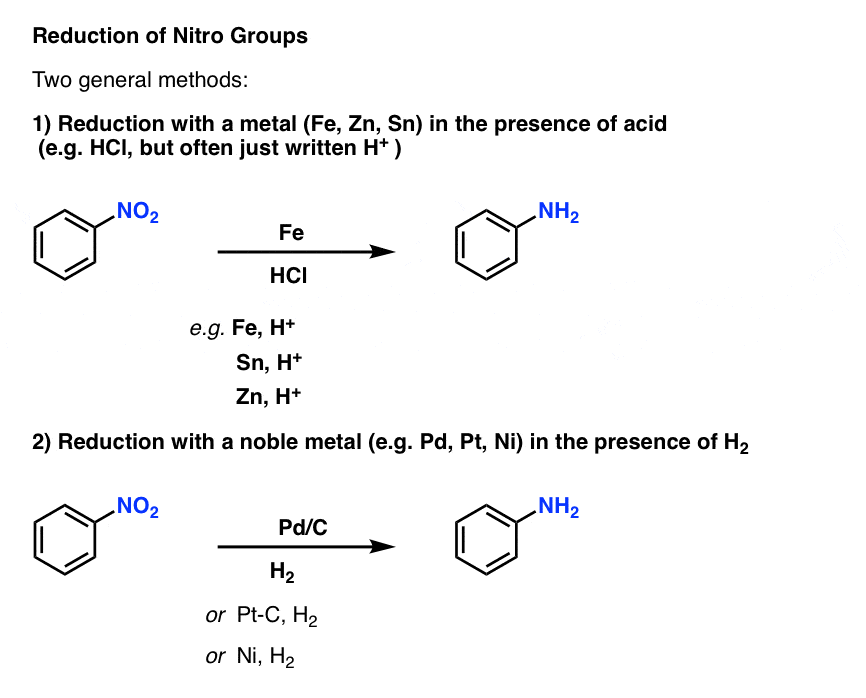
Either method is effective, and can be considered essentially equivalent for our purposes. [What about the mechanism? [Note 1]
One slight advantage of using catalytic hydrogenation is that it can be conducted at neutral pH, and therefore doesn’t affect acid-sensitive functional groups.
3. Protection of the Amino Group
One of the most important features of the reduction of nitro groups to amines is that it converts a strongly deactivating, meta-directing substituent into a strongly activating, ortho-, para- directing substituent.
As it turns out, however, this can actually introduce some new problems!
- First, the amino group is so activating that electrophilic aromatic substitution reactions can occur not just once, but multiple times, resulting in undesired products.
- Secondly, the lone pair on the amine is basic. Reactions that require a Brønsted or Lewis acid catalyst (such as the Friedel-Crafts reactions, sulfonylation, or nitration) don’t end up accelerating the reaction of the electrophile, but instead result in the coordination of the acid to the amine lone pair!
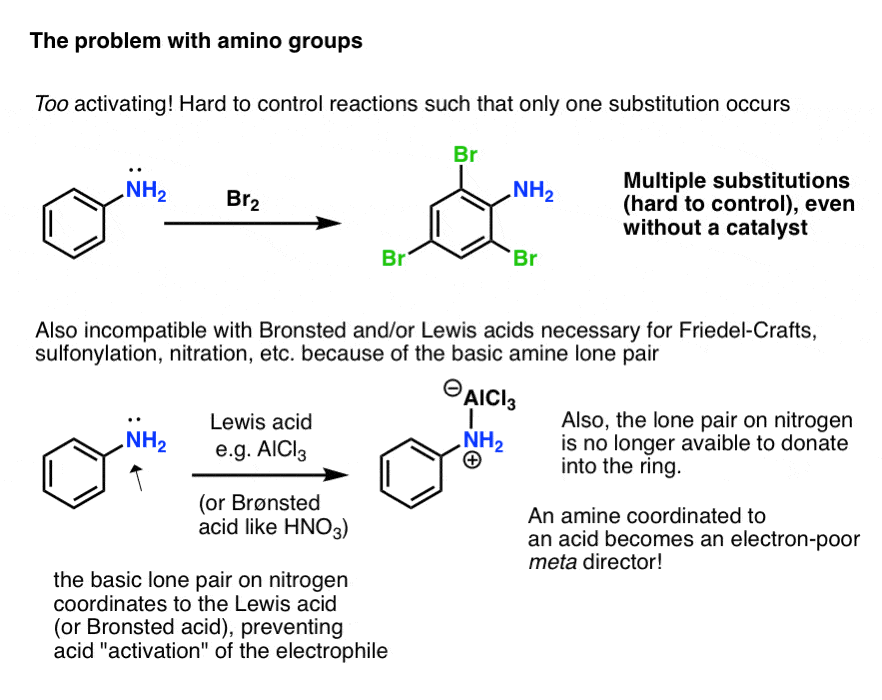
Furthermore, this means that the lone pair on nitrogen is no longer able to donate into the aromatic ring (through “pi donation”). Since nitrogen is more electronegative than carbon, the nitrogen coordinated to an acid is electron-withdrawing and actually behaves as a meta– director!
So how do we tame the “wild horse” that is a free amino substituent?
Fortunately, it’s fairly easy. One common method is to convert the free amine into an amide with a reagent such as acetic anhydride (Ac2O).
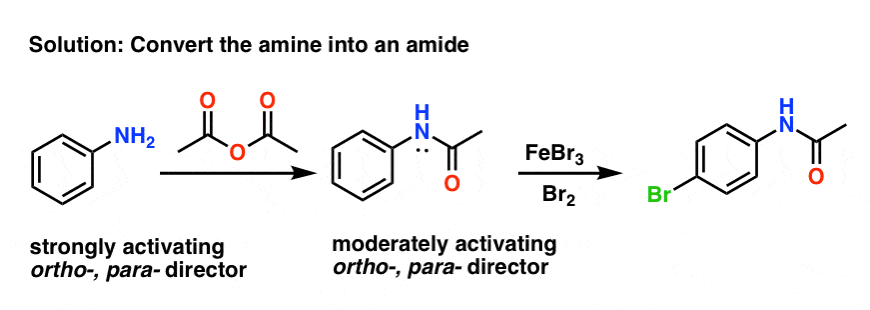
The resulting amide still an ortho- , para– director (note that lone pair on the nitrogen!) but not nearly as activating as the free amine.
Furthermore, amides are much more compatible with Bronsted and Lewis acids than free amines.
If the free amine is desired afterwards, it can be obtained by subjecting the amide to acidic hydrolysis (water, H2SO4, heat).
4. The Baeyer-Villiger Reaction
Another useful reaction for our purposes – and not limited to aromatic ketones – is the Baeyer-Villiger reaction. Treatment of a ketone with a peroxyacid like m-chloroperoxybenzoic acid (m-CPBA) or trifluoroperoxyacetic acid results in a rearrangement reaction where an oxygen atom has been inserted between the carbon of the aromatic ring and the carbonyl carbon, converting the ketone into an ester:
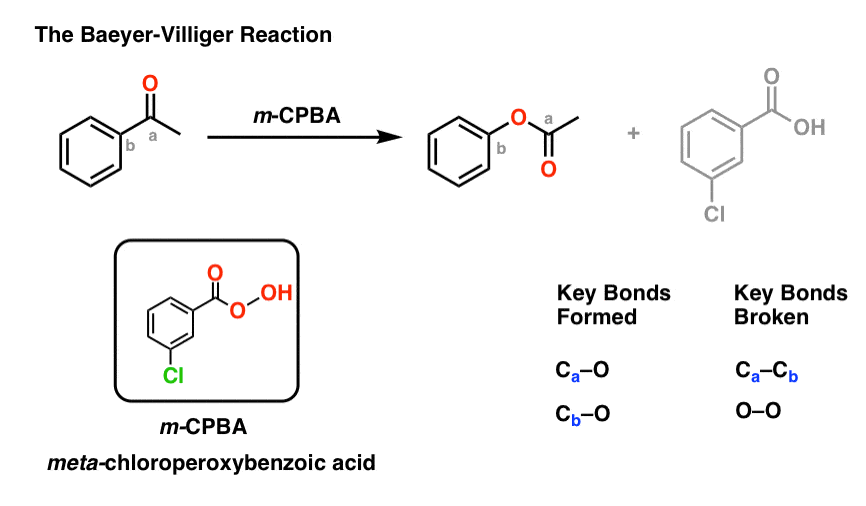
(And no, “typical” oxidants like KMnO4 or chromate or PCC will not do this. There’s a specific reason why peroxyacids work!)
The Baeyer-Villiger reaction fills in an important gap in our “toolbox” of synthetic methods.
Up to this point, we’ve learned how to form C-C, C-N, C-S, and C-(halogen) bonds on an aromatic ring, but not C–O.
Also note that the Baeyer-Villiger, like the reduction of nitro groups to amines, converts a deactivating, meta-directing carbonyl group to an activating, ortho-, para- directing oxygen (those lone pairs on the oxygen are capable of pi-donation).
Now the fun part. How does it work?
5. Mechanism Of The Baeyer-Villiger
In the first step of the Baeyer-Villiger, an oxygen from the peroxyacid attacks the ketone, leading to a tetrahedral intermediate. Proton transfer then leads to the key intermediate:
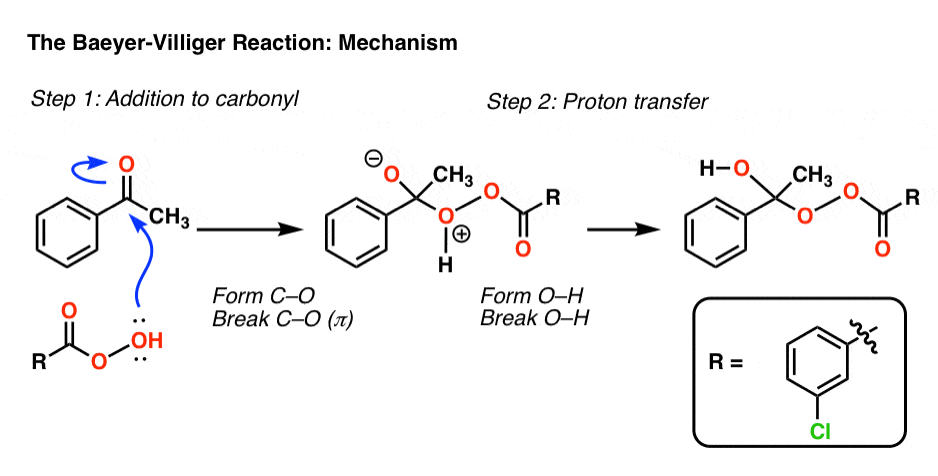
Now comes the key rearrangement step. In this step, a sigma bond acts as a nucleophile, attacking oxygen and breaking the weak (about 35 kcal/mol) O–O bond.
See if you can follow the arrows in this step:
- In the first curved arrow, C–C breaks and C–O forms
- In the second curved arrow, O–O breaks
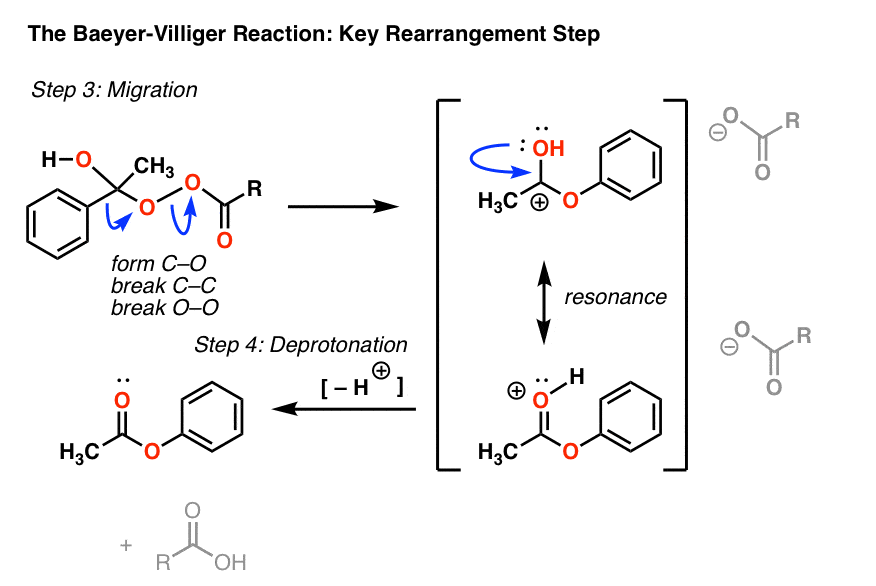
Breaking the C–C bond results in a carbocation. We can then draw a resonance form where a lone pair from oxygen makes a new pi bond with the carbon to give a protonated ester (third curved arrow). [Note – it’s also possible to draw this happening at the same time as the rearrangement, see Note 2 for an example]
This is then deprotonated (by the conjugate base of the acid) to give the final ester product and m-chlorobenzoic acid. (no longer “peroxy”benzoic acid, since it’s lost the O–O bond !)
In rearrangement reactions like this, I find it helpful to draw the “ugly” version of the product first, which has the right connectivity, but drawn poorly. Hover here for image of the “ugly version” (link to image)
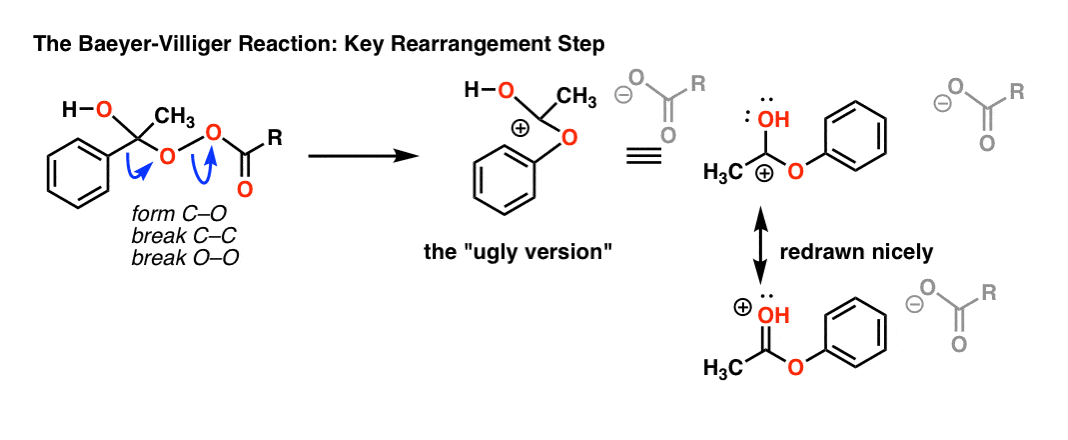{kind=link}
6. Migratory Aptitudes In The Baeyer-Villiger Oxidation
“Why does the phenyl ring migrate to form this ester. Couldn’t the methyl group migrate instead?”
Great question! This is such a great question that answering it will go a little bit beyond the standard course material, so I’ll address this in [Note 3]
To explain in brief, there’s a general trend between migratory aptitude and carbocation stability (with the big exception of phenyl).
An approximate order of migratory aptitude is: tertiary carbon (fastest) > secondary carbon, aryl (e.g. C6H5)> primary carbon > methyl (slowest).
(later in the course (amines), you may encounter the Beckmann, Hofmann, Curtius, and Schmidt rearrangements, which occur through a similar migration step).
Flashback: Hydroboration-Oxidation, And Sigma Bonds As Nucleophiles
Where have we seen a rearrangement step like this before?
Of the three classes of nucleophiles we encounter in organic chemistry [see: The Three Classes of Nucleophiles] (lone pairs, pi bonds, and sigma bonds) sigma bonds as nucleophiles are by far the trickiest to follow. Previously, we’ve seen sigma bonds as nucleophiles in situations like:
- 1,2-hydride shifts (carbocation rearrangements)
- alkyl shifts (carbocation rearrangements)
- the oxidation step in hydroboration
It’s this last example which is most relevant here.
As you may recall, in the oxidation step of hydroboration, the hydrogen peroxide anion attacks boron, creating a tetrahedral compound with a negative charge on boron. Like the Baeyer-Villiger, there is a weak O–O bond which is broken by a migrating sigma bond:
- In the first curved arrow, C–B breaks and C–O forms
- In the second curved arrow, O–O breaks
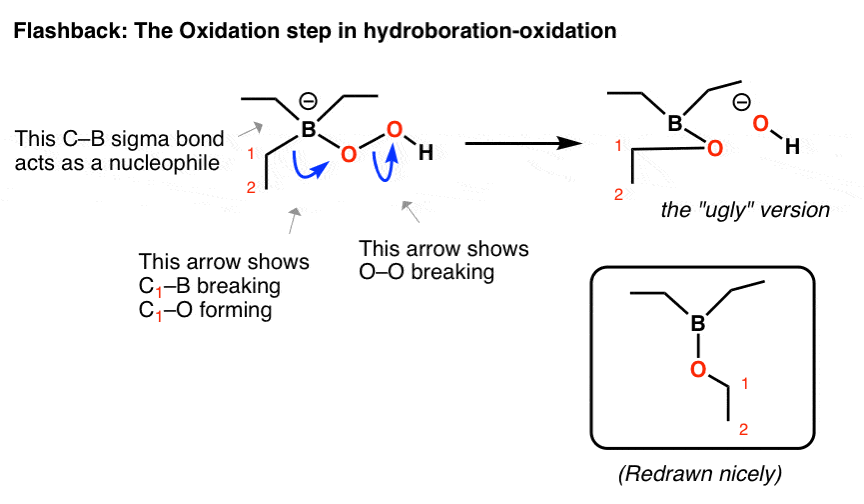
I advise drawing the “ugly” version first to get the connectivity right, and then redraw it nicely.
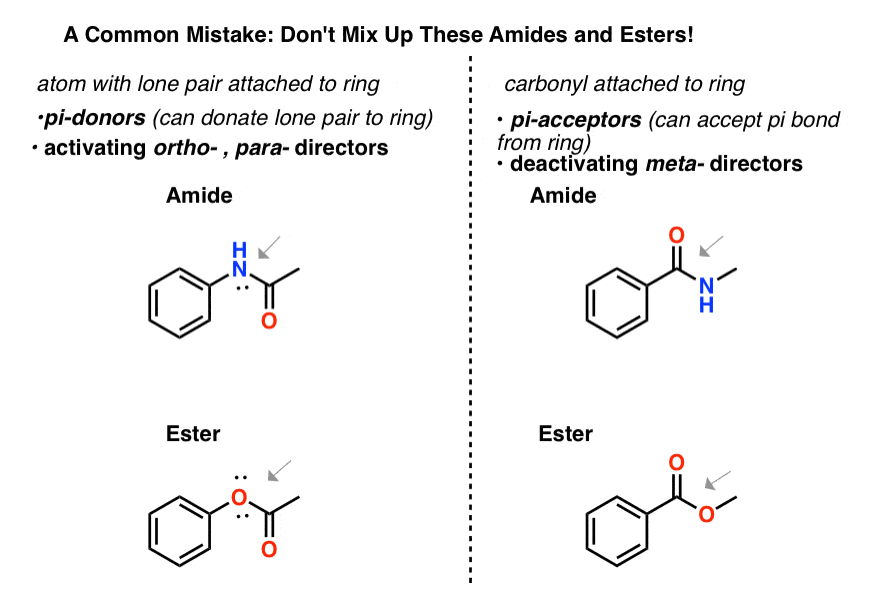
7. A Note of Caution About Mixing Up “Amides” and “Esters”
Make sure you can tell the difference between amides and esters that are ortho-, para- directors, and amides and esters that are meta– directors.
- an atom attached to an aromatic ring that bears a lone pair will be an ortho-, para- director, since it is capable of pi donation into the ring. So an amide with N connected to the aromatic ring or an ester with an O attached to the aromatic ring will be ortho-, para– directors.
- A carbonyl attached to an aromatic ring will be a meta- director, since it can accept a pi bond from the ring. So an amide or ester with the carbonyl attached to the aromatic ring will be a meta director.
Don’t get these two classes of amides and esters mixed up!
8. Summary: Reduction of Nitro Groups and The Baeyer-Villiger Oxidation
Believe it or not, we’ve pretty much come the end of covering substitution reactions on the ring and also reactions of aromatic substituents.
So what now?
It’s time to start putting these reactions together into sequences, and try our hand at the synthesis of aromatic molecules.
With all these reactions in our toolkit, we’ll be able to accomplish quite a lot.
In the next post, the first on aromatic synthesis, we’ll cover some of the basic principles. The second and third posts will cover some synthetic strategies. Finally, we can devote some time to practice problems.
Notes
Note 1. . The mechanism of nitro reduction isn’t generally covered, because it’s loooong (15+ steps), time is limited, and the themes from the mechanism don’t really recur in subsequent parts of the course. Which isn’t to say it isn’t important, just that choices inevitably have to be made, and nitro reduction inevitably ends up on the cutting room floor. Here’s roughly what the reaction would look like with Sn and acid hover for popup image (link to image)
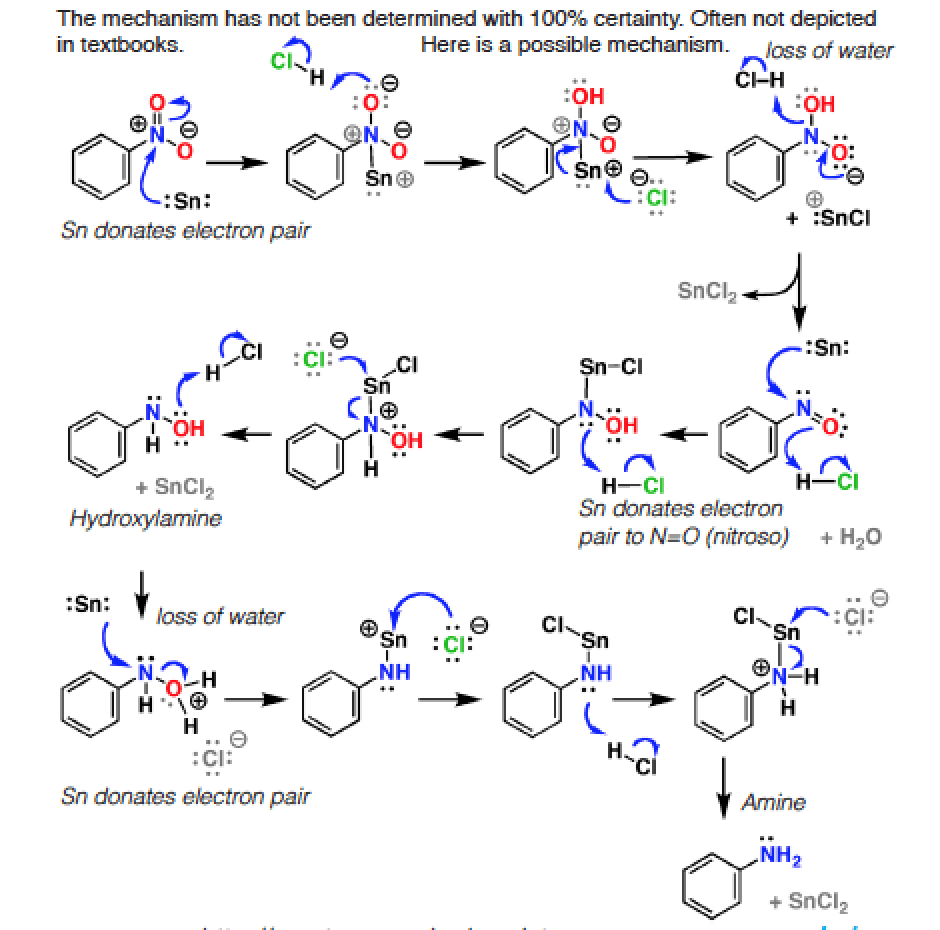{kind=link}
Note 2. The C-O (pi) bond formation is technically just a resonance form , so one could alternatively draw it as a concerted process. Hover for image of C-O pi bond participating [(link to image)
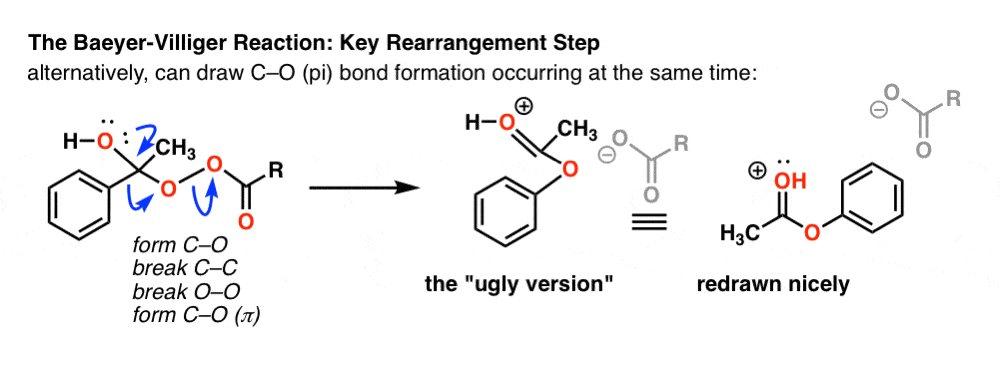{kind=link}
Note 3. The Baeyer-Villiger And Migratory Aptitude
OK, this is pretty far beyond introductory course stuff, so feel free to skip. Still, the question remains: why the weird order of migratory order in the Baeyer-Villiger?
tertiary > secondary, aryl > primary > methyl
It (mostly) goes in the familiar order of carbocation stability (tertiary > secondary > primary > methyl) except that aryl is intermediate between secondary and primary. By now we’ve seen lots of examples that show conclusively that aryl carbocations are more unstable than primary carbocations. So what gives?
Our best hypothesis is that it doesn’t actually go through a direct migration of the C-C sigma bond, but in fact proceeds through attack of the oxygen by the pi system of the aromatic ring. This results in a delocalized carbocation (an “arenium ion”) highly stabilized by resonance, which quickly breaks down to restore aromaticity en route to the final ester product:
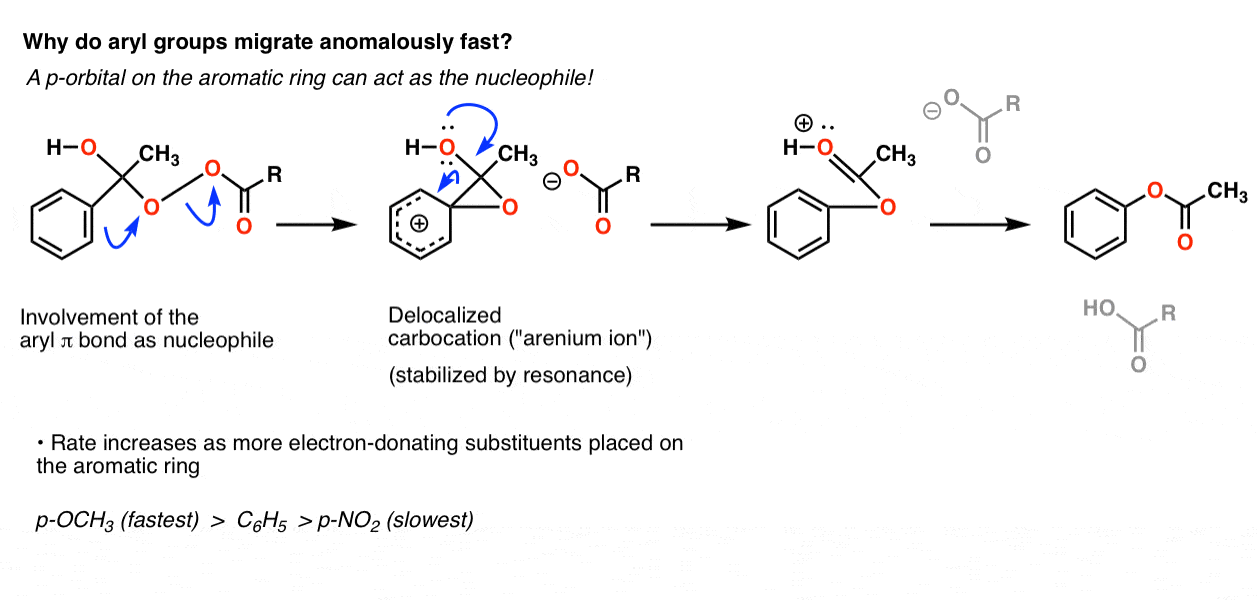
As we’d expect from this mechanism, electron-donating groups on the aromatic ring (such as O–CH3) speed the reaction relative to C6H5 itself, and electron-withdrawing groups (such as NO2) retard the rate.
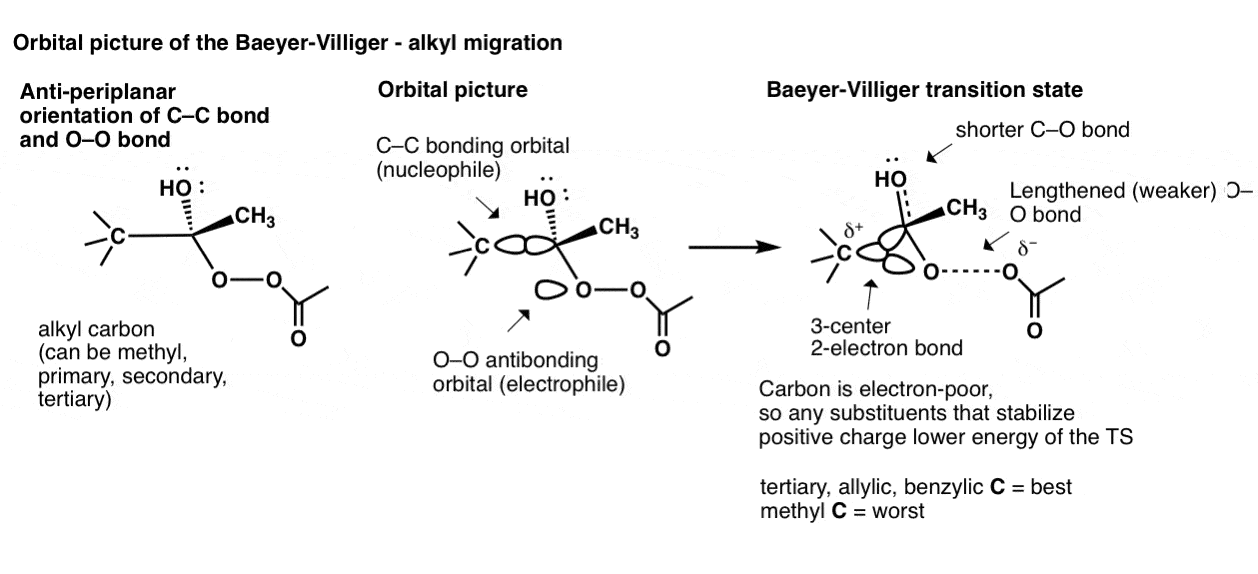
To go even deeper we can look at the orbital picture of an alkyl migration in the Baeyer-Villiger. This begins with the C–C bond aligned in an anti-periplanar arrangement with the O–O bond. The C–C bonding orbital then begins overlap with the O–O antibonding orbital, leading to a transition state where there are three atoms sharing two electrons! (This is called, not surprisingly, a “3-center, 2-electron bond”.
The stability of this transition state is directly related to how well the three groups on the alkyl carbon stabilize positive charge. Hence, tertiary (as well as allylic and benzylic) carbons migrate fastest, followed by secondary, primary, and methyl.
The migration of an aryl group involves more than just the C–C sigma bond; the p-orbital on the carbon can get involved as well!
So what happens in this transition state is that the p-orbital donates into the O–O antibonding orbital, leading to a transition state where the positive charge is spread out over the five carbons in the aromatic ring; not a 3-center, 2-electron bond, as before.
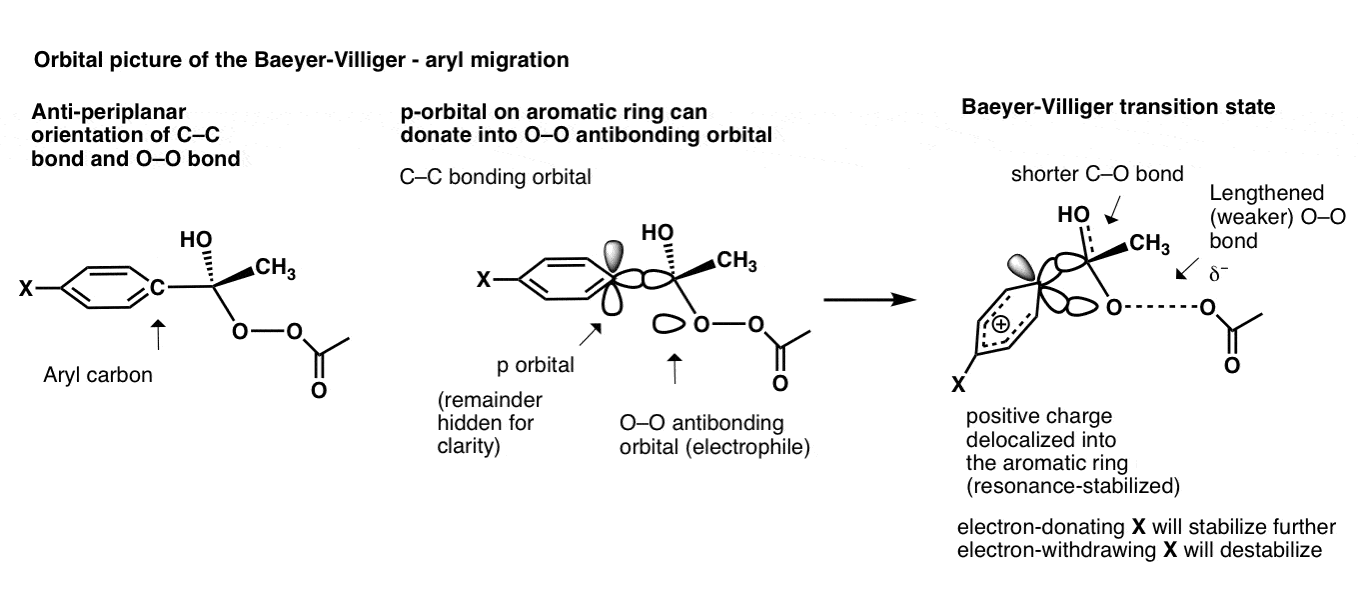
As you can imagine, electron donating groups “X” such as OCH3 stabilize the transition state (increasing the rate) and electron-withdrawing groups such as NO2 destabilize the transition state (slowing the rate).
Theoretical organic chemist Henry Rzepa has a useful blog post discussing the mechanism of the Baeyer-Villiger (particularly the proton-transfer step) here.
Quiz Yourself!
Become a MOC member to see the clickable quiz with answers on the back.
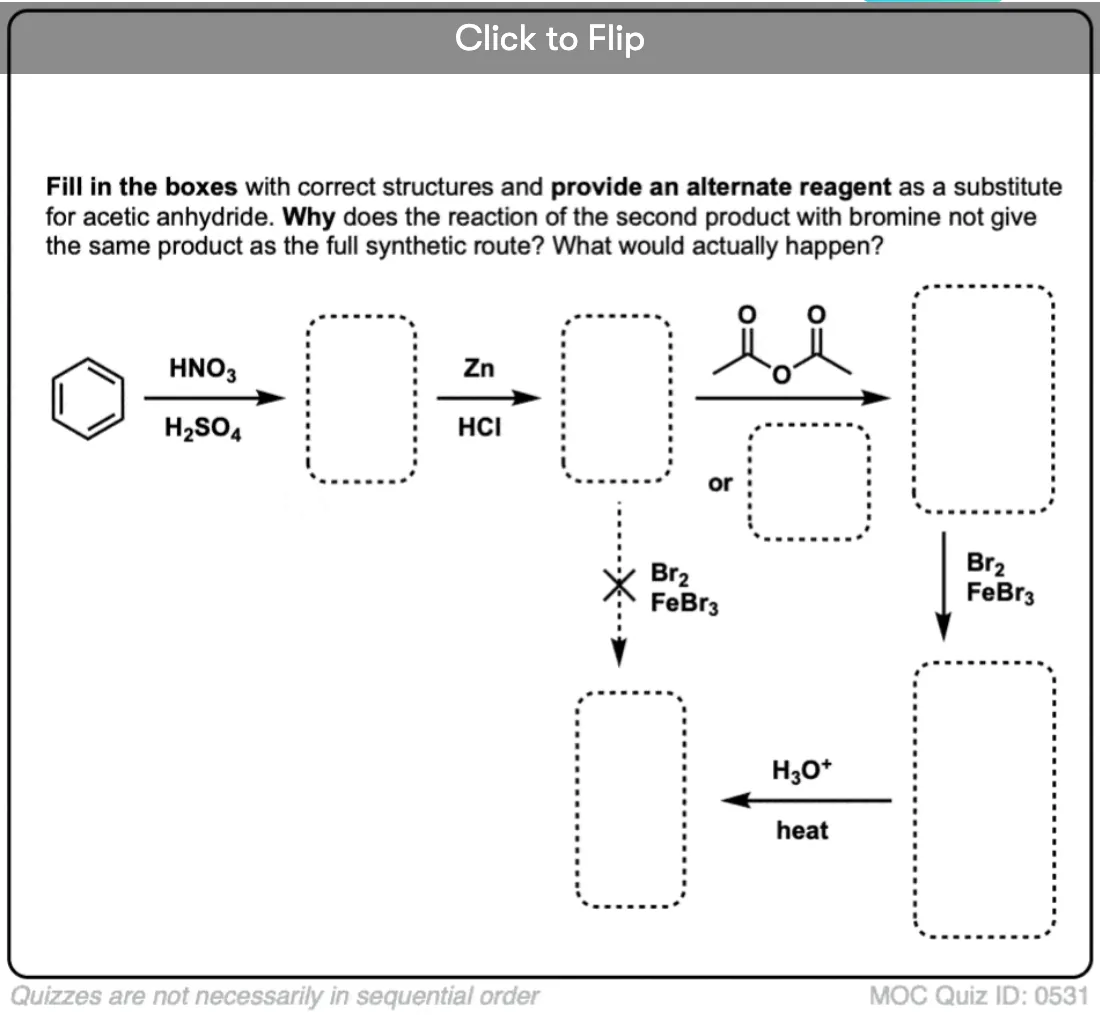
Become a MOC member to see the clickable quiz with answers on the back.
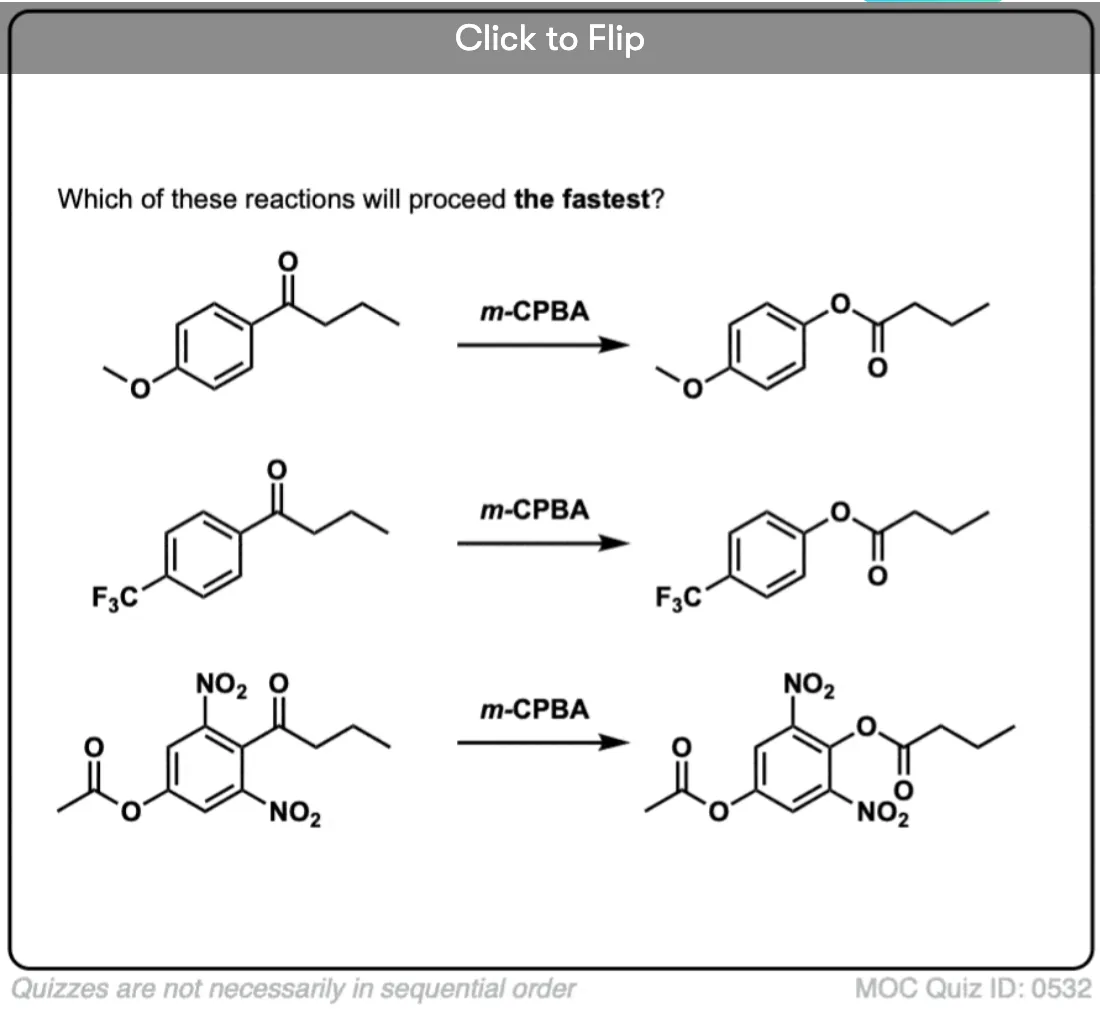
Become a MOC member to see the clickable quiz with answers on the back.
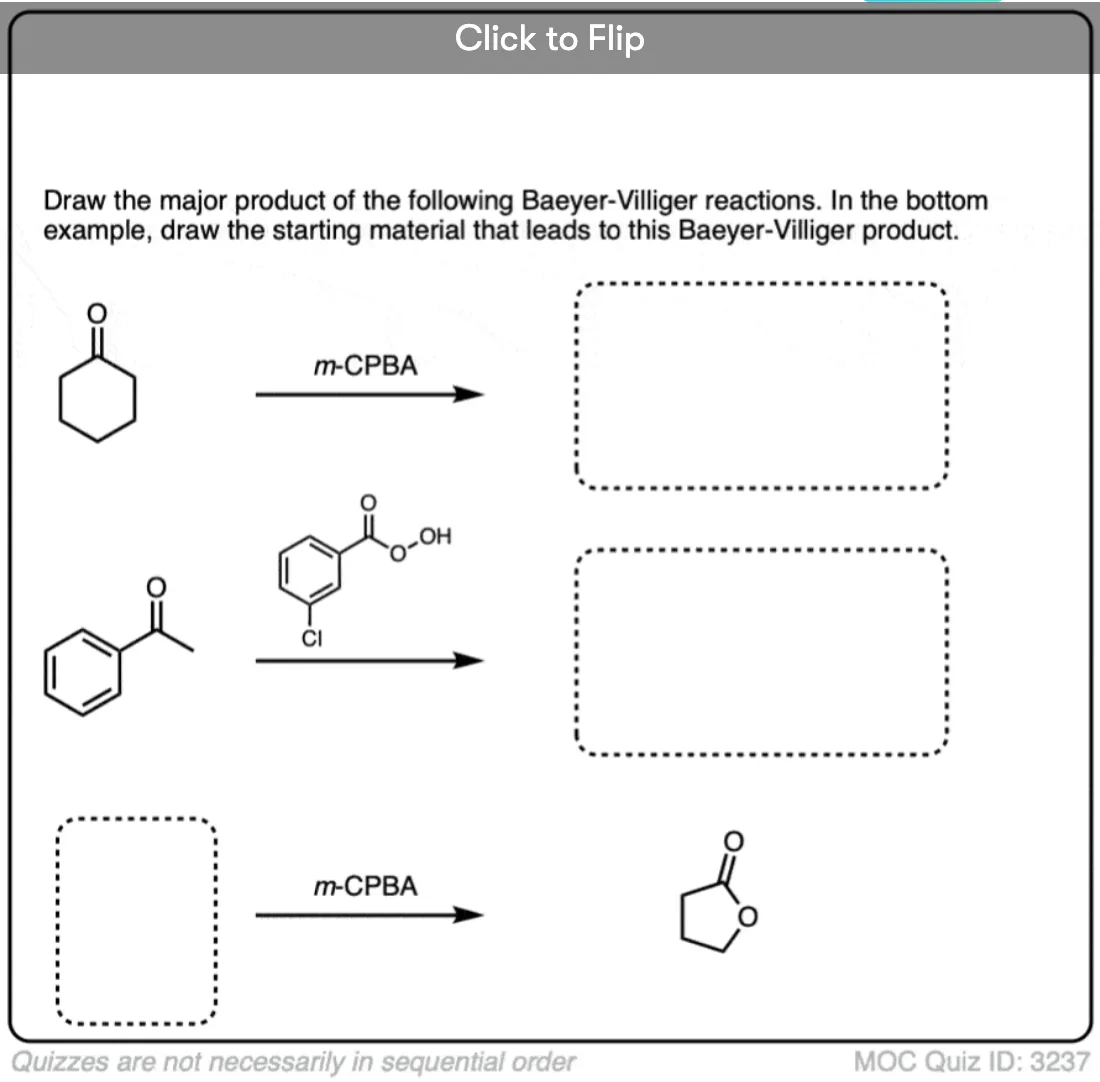
Become a MOC member to see the clickable quiz with answers on the back.
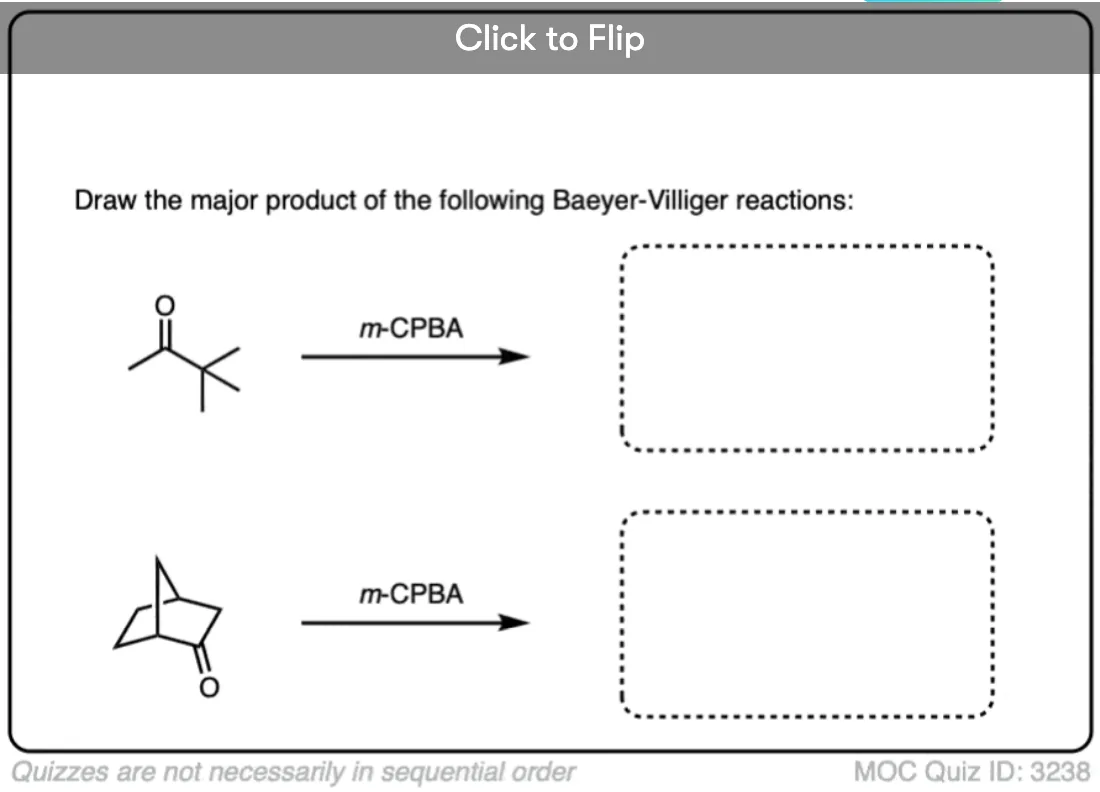
Become a MOC member to see the clickable quiz with answers on the back.
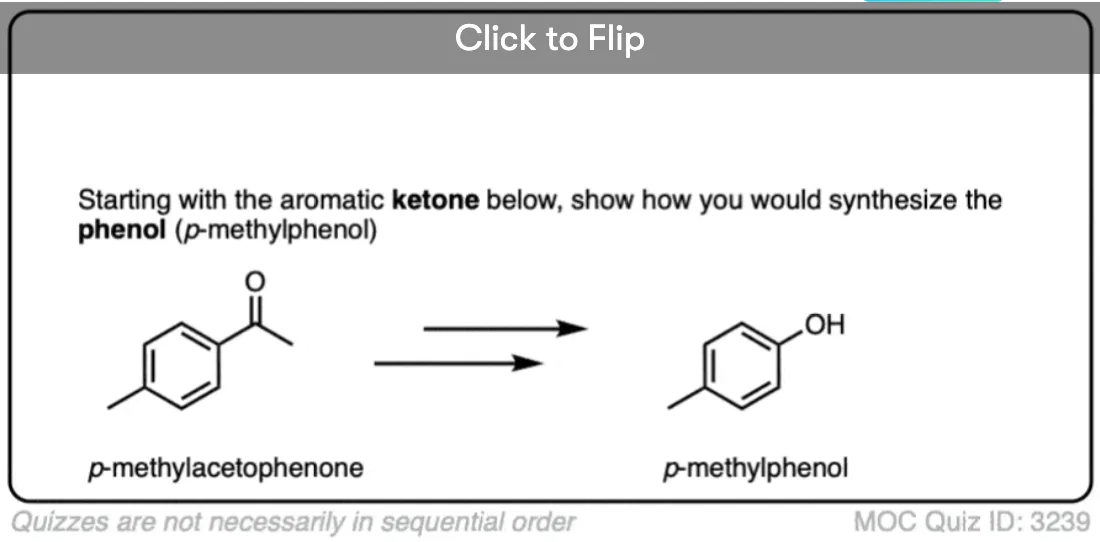
Become a MOC member to see the clickable quiz with answers on the back.
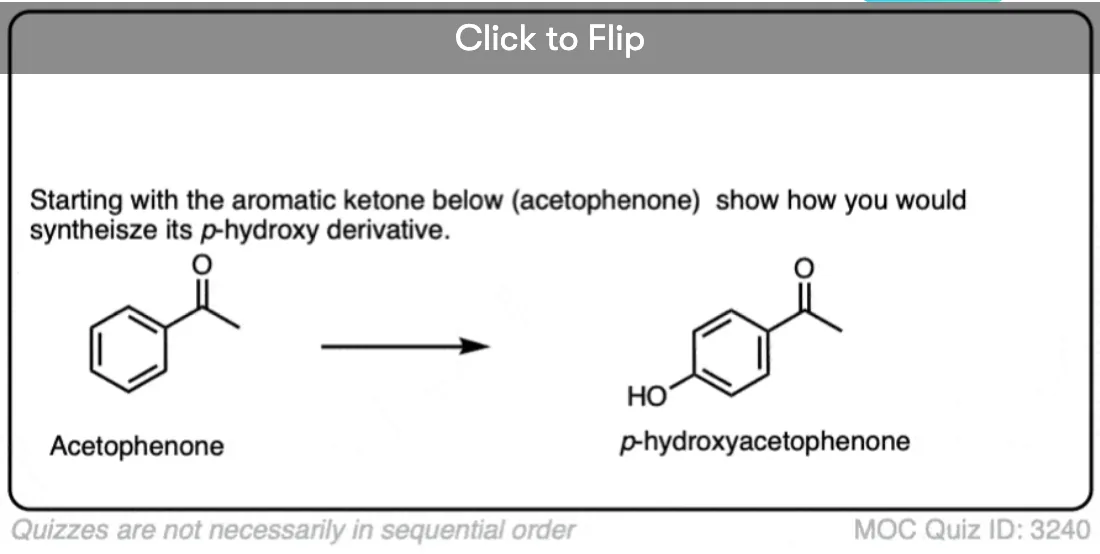
Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading
Baeyer-Villiger oxidation:
- Einwirkung des Caro’schen Reagens auf Ketone
Adolf von Baeyer, Victor Villiger
Ber. 1899, 32 (3), 3625
DOI: 10.1002/cber.189903203151
This paper by Nobel Laureate Adolf von Baeyer first describes what is now known as the Baeyer-Villiger rearrangement, using a mixture of sodium persulfate and concentrated sulfuric acid (Caro’s acid). - 23. Synthetic approaches to .alpha.-methylene-.gamma.-lactones via cycloadditions of ketenes
Alfred Hassner, Harold W. Pinnick, and Jay M. Ansell
The Journal of Organic Chemistry 1978 43 (9), 1774-1776
DOI: 10.1021/jo00403a032
This paper has a representative procedure for a Baeyer-Villiger oxidation in the experimental section. - 100 Years of Baeyer–Villiger Oxidations
Michael Renz and Bernard Meunier
Eur J. Org. Chem. 1999, 4, 737
DOI: 1002/(SICI)1099-0690(199904)1999:4<737::AID-EJOC737>3.0.CO;2-B
This review on the Baeyer-Villiger oxidation includes a detailed historical perspective on the development and history of the reaction. - The Baeyer–Villiger Oxidation of Ketones and Aldehydes
Krow, G. R.
Org. React. 1993, 251
DOI: 10.1002/0471264180.or043.03
This long, detailed review includes an in-depth discussion of the mechanism, substrate scope, limitations, and experimental procedures for the Baeyer-Villiger oxidation. - The Baeyer−Villiger Reaction: New Developments toward Greener Procedures
-J. ten Brink,I. W. C. E. Arends, and, and R. A. Sheldon
Chemical Reviews 2004 104 (9), 4105-4124
DOI: 10.1021/cr030011l
This review gives a modern perspective on the Baeyer-Villiger oxidation and describes procedures using more “environmentally friendly oxidants (e.g. O2). - The Baeyer-Villiger Oxidation of Aromatic Aldehydes and Ketones with Hydrogen Peroxide Catalyzed by Selenium Compounds. A Convenient Method for the Preparation of Phenols
Ludwik Syper
Synthesis 1989, 3, 167-172
DOI: 1055/s-1989-27183
Migration of aryl over H does rarely happen, as seen in some of the examples here. - Experimental Support for the Primary Stereoelectronic Effect Governing Baeyer−Villiger Oxidation and Criegee Rearrangement
Richard M. Goodman and Yoshito Kishi
Journal of the American Chemical Society 1998, 120 (36), 9392-9393
DOI: 10.1021/ja982188g
An short communication in which some substrates used for B-V oxidation yield products demonstrating that stereoelectronic effects are the reason for migratory aptitude. Prof. Yoshito Kishi is thought of as the philosophical successor to the legendary chemist Prof. R. B. Woodward – he inherited Woodward’s research group after Woodward passed away, and continued Woodward’s legacy of challenging total syntheses, culminating in the synthesis of the marine natural product palytoxin, considered the most challenging synthesis to date. Prof. Kishi also has his name associated with a reaction, the Nozaki-Hiyama-Kishi reaction, the mechanism of which was discovered rather serendipitously.Reduction of aromatic nitro groups:
A large variety of methods are available to do this reduction, which can take place either by hydrogenation or using a metal in acid. - 2-AMINO-p-CYMENE
F. H. Allen and James VanAllan
Org. Synth. 1942, 22, 9
DOI: 10.15227/orgsyn.022.0009
An early example of this reduction in Organic Synthesis, a source of reliable and independently-tested synthetic organic laboratory procedures. This uses hydrogenation with Raney Ni to reduce the nitro group. - Reduction of Organic Compounds by Lithium Aluminum Hydride. III. Halides, Quinones, Miscellaneous Nitrogen Compounds
Robert F. Nystrom and Weldon G. Brown
Journal of the American Chemical Society 1948, 70 (11), 3738-3740
DOI: 1021/ja01191a057
LiAlH4 can also be used for reducing aromatic nitro groups, but this will give azobenzenes. This seems to be a very convenient method for azobenzene synthesis, as in the case of p-nitrobromobenzene – “The product from the reduction (at room temperature) of p-nitrobromobenzene, 4,4′-dibromoazobenzene, being insoluble in water and only slightly soluble in ether, separated in crystalline form upon the addition of dilute sulfuric acid. It was obtained in very pure form merely by filtration and washing with hot water.” - Selective reduction of aromatic nitro compounds with stannous chloride in non acidic and non aqueous medium
D. Bellamy, K. Ou
Tet. Lett. 1984, 25 (8), 839-842
DOI: 10.1016/S0040-4039(01)80041-1
SnCl2 in ethanol can be used as a pH-neutral, nonaqueous system for nitro reduction. - Reduction of aromatic nitro compounds by secondary alcohols using rhodium complexes as catalysts
F. Liou and C. H. Cheng
The Journal of Organic Chemistry 1982, 47 (15), 3018-3021
DOI: 10.1021/jo00136a046
Among the hundreds of variants of this reaction that are known, Rh complexes can be used as homogenous catalysts for the reduction of nitro groups by transfer hydrogenation.
Thank you for your help for giving such detailed things
James, just one little coment. In the second question in quiz, you should mentioned that besides uncontrolable orto/para alkylation of aniline also meta-alkyl aniline is formed (according to disadvantages of using free amine group under acidic conditions).
If i am not wrong the migratory aptitude is H (fastest)> phenyl group > tertiary > secondary > primary (slowest)
Isn’t it?
From March / Smith, Advanced Organic Chemistry 5th ed. “For unsymmetrical ketones the approximate order of migration is tertiary alkyl > secondary alkyl and aryl > primary alkyl > methyl.
I would put H before tertiary alkyl, although apparently migration of aryl over H does happen (rarely). https://www.thieme-connect.com/products/ejournals/abstract/10.1055/s-1989-27183