Home / Common Mistakes with Carbonyls: Carboxylic Acids… Are Acids!
Organic Chemistry Tips and Tricks
Common Mistakes with Carbonyls: Carboxylic Acids… Are Acids!
Last updated: January 23rd, 2024 |
Carboxylic acids… are acids.
I know that seems obvious. But it’s a near certainty that students taking Org 2 for the first time will forget this occasionally.
Here are two common mistakes that I see *all the time*.
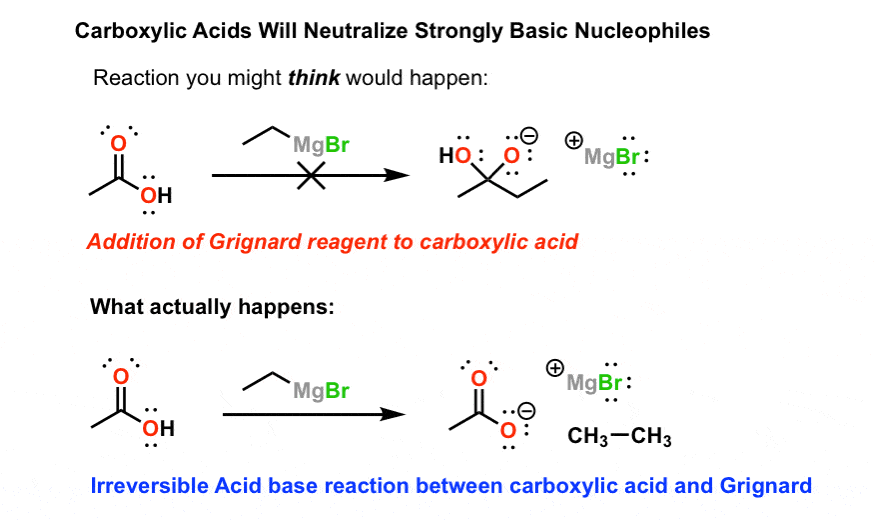
1) Reactions of Grignard reagents with carboxylic acids.
Grignard reagents (with the general structure RMgBr) are great nucleophiles. They add to ketones, aldehydes, esters (twice), acid halides (twice), epoxides, and a number of other carbonyl-containing compounds.
For students getting their feet wet with carbonyl chemistry, it can be tempting to also draw Grignard reagents adding to carboxylic acids.
They don’t.
That’s because carboxylic acids are… acids, and Grignard reagents are very strong bases. So instead of adding to the carbonyl carbon, the Grignard is simply protonated first. And the resulting conjugate base of the carboxylic acid (a carboxylate) is too unreactive to react further.
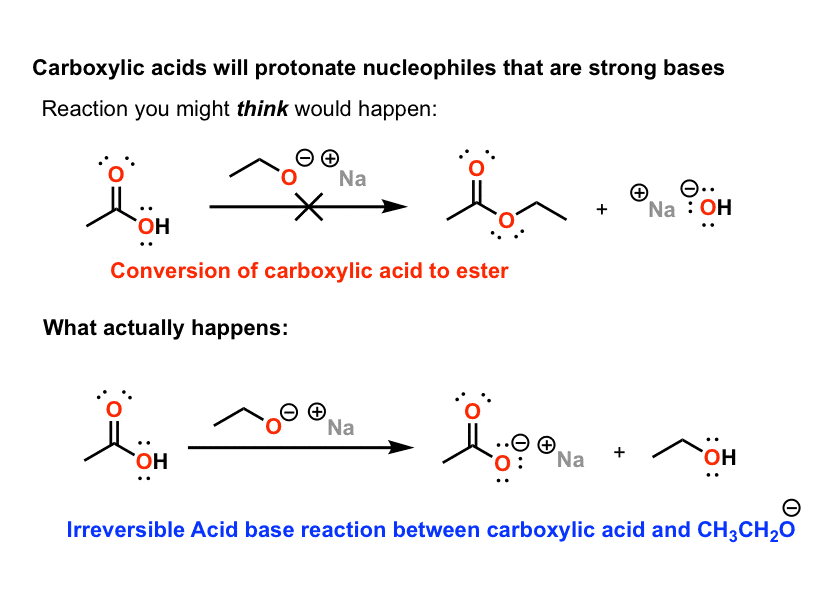
2) Reaction with alkoxides (deprotonated alcohols)
Carboxylic acid derivatives like esters, anhydrides, and acid halides react well with good nucleophiles like HO- and RO- . The pattern becomes familiar quite quickly: 1,2 addition, followed by 1,2 elimination. Seeing this pattern, students get lulled into a false sense of security that carboxylic acids will react this way as well.
They don’t – for the same reasons that Grignard reagents don’t. Carboxylic acids are acids. They protonate strong bases (such as alkoxides) and leave behind the carboxylate, which – again – is unreactive.
It seems silly to repeat this a third time, but it happens *all the time*. You might not think you will do this. Chances are, at some point, you will. It’s an easy mistake to make. So let’s say it one last time:
Carboxylic acids…. are acids!
———-
Note below: It’s a pretty good rule of thumb to assume that acid-base reactions will happen faster than reactions that involve nucleophilic attack, such as addition to carbonyls. If you’re keen, there’s a name for this: the principle of least motion. That’s why we see protonation of the Grignard reagent before it has time to add to the carbonyl carbon.
00 General Chemistry Review
01 Bonding, Structure, and Resonance
02 Acid Base Reactions
03 Alkanes and Nomenclature
04 Conformations and Cycloalkanes
05 A Primer On Organic Reactions
06 Free Radical Reactions
07 Stereochemistry and Chirality
08 Substitution Reactions
09 Elimination Reactions
10 Rearrangements
11 SN1/SN2/E1/E2 Decision
12 Alkene Reactions
13 Alkyne Reactions
14 Alcohols, Epoxides and Ethers
15 Organometallics
16 Spectroscopy
17 Dienes and MO Theory
18 Aromaticity
19 Reactions of Aromatic Molecules
20 Aldehydes and Ketones
21 Carboxylic Acid Derivatives
22 Enols and Enolates
23 Amines
24 Carbohydrates
25 Fun and Miscellaneous
26 Organic Chemistry Tips and Tricks
- Common Mistakes: Formal Charges Can Mislead
- Partial Charges Give Clues About Electron Flow
- Draw The Ugly Version First
- Organic Chemistry Study Tips: Learn the Trends
- The 8 Types of Arrows In Organic Chemistry, Explained
- Top 10 Skills To Master Before An Organic Chemistry 2 Final
- Common Mistakes with Carbonyls: Carboxylic Acids... Are Acids!
- Planning Organic Synthesis With "Reaction Maps"
- Alkene Addition Pattern #1: The "Carbocation Pathway"
- Alkene Addition Pattern #2: The "Three-Membered Ring" Pathway
- Alkene Addition Pattern #3: The "Concerted" Pathway
- Number Your Carbons!
- The 4 Major Classes of Reactions in Org 1
- How (and why) electrons flow
- Grossman's Rule
- Three Exam Tips
- A 3-Step Method For Thinking Through Synthesis Problems
- Putting It Together
- Putting Diels-Alder Products in Perspective
- The Ups and Downs of Cyclohexanes
- The Most Annoying Exceptions in Org 1 (Part 1)
- The Most Annoying Exceptions in Org 1 (Part 2)
- The Marriage May Be Bad, But the Divorce Still Costs Money
- 9 Nomenclature Conventions To Know
- Nucleophile attacks Electrophile
Well, I might not heard the definitive answer here, but anyway I’ll share the answer provided by the maître Grignard himself.
In short – yes, reaction of pyruvate (ester) with 1 mole equivalent of Grignard reagent will result in a-substituted lactate.
More generally, a-ketoacid esters will give α-substituted hydroxyacid esters. Or bi-tertiary glycols, when 3 mole equivalents of Grignard reagent are used (tertiary hydroxy groups stemming from carbonyl and carboxyl group respectively). Yields are low, in the 25-35% range.
β-ketoacids provide basically nothing useful, due to the acidity of their α-protons.
γ-ketoacids provide upon workup γ-substituted γ-lactones, also with yields around 30%.
Also, apparently ketoacid salts, despite their low solubility in ethers, work as (in)effectively as esters.
Cited literature:
-V. Grignard, Action des combinaisons organomagnésiennes mixtes sur les èthers d’acides cètoniques (II), Comptes rendus, t. 135 (1902), p. 627-630.
-V. Grignard, Action des combinaisons organomagnêsiennes
sur les èthers β-cétoniques, Comptes rendus, t. 134 (1901), p. 849-851.
-P. K. Porter, The Action of the Grignard Reagent on Keto Acids, J. Am. Chem. Soc. 1923, 45, 4, 1086–1087, DOI 10.1021/ja01657a501
-W. A. Noyes, C. S. Marvel, CYANCARBOXETHYL 3,3-DIMETHYL CYCLOPENTANONE, J. Am. Chem. Soc. 1917, 39, 6, 1267–1271, DOI 10.1021/ja02251a015
-D. T. Jones, G. Tattersall, CLXX.—A new synthesis of isocaprolactone and certain derivatives, J. Chem. Soc., Trans., 1904,85, 1691-1694, DOI 10.1039/CT9048501691
That is extremely useful. Love the references. Thank you!
I wasn’t thinking of any particular reagent, just a generic alkyl (like ethyl) magnesium bromide. I was interested mainly because I couldn’t find anything in my books about this case. Only general reactions with aldehydes, ketones, etc. and a reminder that reagents with an acidic enough hydrogen would protonate the Grignard, no mentions about salts, keto acids and other such niche cases.
I’d be grateful if you could direct me towards some articles/literature on this topic.
You mentioned that the deprotonation of a methyl group could be a concern – would the adjacent carbonyl increase methyl’s hydrogens’ acidity that much? I knew that Grignards are strong bases capable of deprotonation of alkynes, but not to such a degree.
If you’re asking about the Grignard’s identity, would that mean that different reagents (like phenyl magnesium bromide) could react differently? In what ways? Sorry if I sound demanding, apparently I stumbled upon an interesting topic and I’d love to learn more.
Apologies if this comment does not land within the original thread, but the site would not allow me to reply otherwise.
Hello. I greatly enjoy your website.
Would there be a reaction between a Grignard reagent and a ketocarboxylate, i.e. salt of a ketoacid, like sodium pyruvate? Would the product be an alpha-alkylated lactate?
Thank you in advance.
It’s possible. Deprotonation of the methyl group of pyruvate might be a concern, but likely some addition would occur. What’s the identity of the Grignard reagent?
My confusion is now totally wiped out
😃…
Making Alcohol from Grignard reagent
I was too confused while solving questions. This post made it wayy clear.
Thanks.
A carboxylate actually has the charge delocalized between the two oxygens, This might explain why it isn’t keen on behaving like a carboxyl compound.
Yes, exactly. The negatively charged oxygen is a strong pi donor and the carbonyl carbon is less electrophilic for this reason.
addition of nucleophile is not possible with HCOO- why? even it does not react with grignard reagent
Thanks
Could you explain why HBr doesn’t add to a carbonyl in a nucleophilic addition??
The C-Br bond is much weaker than the C-O pi bond. Even if a C-Br bond did form, it would quickly be expelled to regenerate the carbonyl.
Please explain: Grignard reagent + carboxylic acid chloride –> ketone + Mg-halides.
Is the C-Cl bond so reactive as to preclude simple protonation of said Grignard?
By “carboxylic acid chloride” it’s meant, “acid chloride” which in fact does not contain an acidic hydrogen. So protonation of the Grignard is not an issue here, although it would be with carboxylic acids.
what other functional groups can cause a similar problem ?
Hydroxyl groups (OH), thiols (SH), and generally any other mildly acidic functional groups. Carboxylic acids are by far the most common situation one encounters.
Will grignards reagent react with the acidic alpha hydrogen next to C=O in RCOCl?
Generally not, unless the acid chloride is extraordinarily hindered. C-H bonds don’t deprotonate as quickly as O-H bonds since 1) there has to be reorganization (rehybridization) of the bonds, and 2) the acidity of C-H is dependent on its alignment with the carbonyl.
Great explanation!
thank you! my doubts are solved :)
This is Matt, I’m a tutor that works through Master Organic. To answer your question frank, I will start by agreeing that it does seem a bit crazy at first glance that a grignard will simply deprotonate a carboxylic acid while the the alkyl lithium irreversibly forms a dianion through sequential deprotonation/addition. In most context grignards and alkyl lithiums react in the same way, although alkyl lithiums are more often used when deprotonation is the specific goal, I have noticed (ylide formation, for example). If you study enough mechanisms you will eventually come upon many of these frustratingly inconsistent examples that must be rationalized by some hand waving. It’s possible that nobody knows the answer for sure it probably has something to with either:
1) Slightly stronger lewis acidity of Li+ cation in this context
2) Slightly higher nucleophilicity of the C-Li bond compared to the C-Mg bond
This may be an oversimplification though, and I welcome anyone who knows of experimental data or computational studies that explain this. The mechanism itself could be entirely different, as these reagents do sometimes do funky things such as single electron transfer (SET) radical mechanisms.
Matt – I would say your answer (1) is good, but your answer (2) is redundant. By definition, if the R-Li is more reactive it is because it means it has “higher nucleophilicity.”
But back to the Li Lewis acidity. You can compare it to the difference between LiAlH4 and NaAlH4 or NaBH4 and LiBH4. In both cases, the Li salts are more reactive. A lot of the difference is likely due to the Lewis acidity, but there could also be solvation differences that affect reactivity as well. In fact, solvation/Lewis acidity pretty much accounts for the difference in reactivity between LAH and NaBH4. Thermochemical studies of the bare ions AlH4- and BH4- show that there is no difference in the hydride binding energies, and the activation energies for reduction of CO2 in the absence of solvent and counter-ion are the same for AlH4- and BH4-.
It’s not always easy to think about the structures of things like organometallics or inorganic salts in organic solution. Even subtle differences in structure like diethyl ether and THF can change solvation properties, and these things can have large effects on reactivity. Fortunately, we have folks like Hans Reich and Dave Collum to help us understand it better.
Hi James!
Great post, I had understood it when I saw it last semester but recently I came across that Organolithium reaction you had mentioned in the comments. I’m a little confused as to why if you add the 2 equivalents of Organolithium you can use the first to deprotonate and the 2nd to add on but you can’t do the same thing with 2 equivalents of Grignard reagents. Is it simply that the Grignard is weaker? I had always thought the two were extremely powerful nucleophiles/bases and didn’t differ too much.
-Confused But Would Love To Be Enlightened (Frank W.)
Ahh..that embarrassing feeling when you realize that you forgot what you know oh so well just a few days ago.
thanks that’s a lot of help so how could you prepare an ester from a carboxylic acid without carrying out fischer esterification?
Carboxylic acids + amines is another common proton transfer example that students miss. I don’t know how many RC(=NR)OH products I’ve seen – it’s a lot.
thanks it help me so much
how most grinard react with carb. acid?
As shown here, they are usually protonated!
Tanks, i was so confused
So is there a way to add alkyl groups to carboxylic acids? Lets say I wanted to go from a carboxylic acid to a tertiary substituted alcohol.
If you use organolithium reagents you can add alkyl groups to carboxylic acids to form ketones. These will also deprotonate the carboxylic acid to the carboxylate, but are powerful enough to add to subsequently form the C-C bond. However I believe that the intermediate that is formed is fairly stable and will not eliminate to give the ketone until workup. To get the tertiary alcohol you’d have to submit the ketone to another equivalent of organolithium (or Grignard if you prefer). Hope this answers your question.
Thanks!!
I was also confused this matters.
Now I got it! :)