Aldehydes and Ketones
All About Enamines
Last updated: April 16th, 2025 |
Enamines – formation, properties, reactions, and mechanisms.
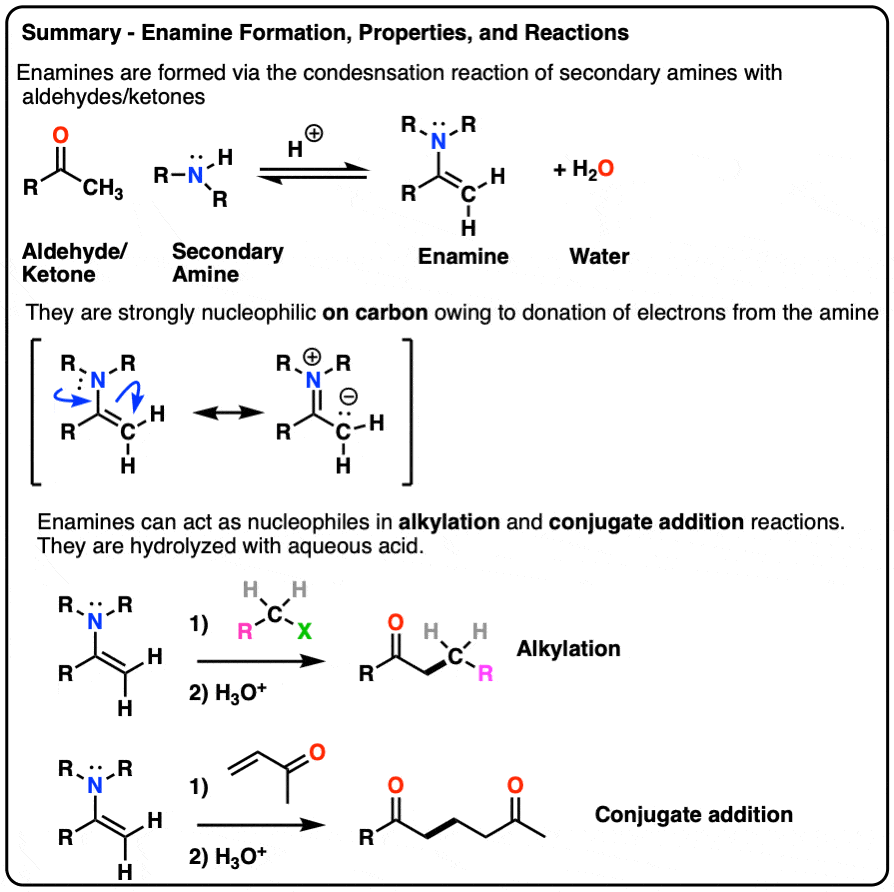
- Enamines are formed from the reaction of a secondary amine with an aldehyde or ketone.
- Because they have a significant resonance form where there is a negative charge on the alpha carbon, they are nucleophilic.
- Enamines undergo reactions with electrophiles such as alkyl halides and Michael acceptors
- They are hydrolyzed with strong acid.
Note – this is a significant update of an older post on imines and enamines.
Table of Contents
- What Are Enamines And How Are They Made?
- Mechanism for Enamine Formation
- Properties of Enamines (Hint: They’re Nucleophiles)
- Properties of Enamines – Basicity and Geometry
- Reactions of Enamines: Alkylation
- Reactions of Enamines: Michael Addition (Conjugate Addition)
- Reactions of Enamines: Hydrolysis With Aqueous Acid
- Summary – Enamine Properties and Reactivity
- Notes
- Quiz Yourself!
- (Advanced) References and Further Reading
1. What Are Enamines And How Are They Made?
The previous post in this series covered imines, the nitrogen-containing cousins of aldehydes and ketones that are obtained through treatment of an aldehyde or ketone with a primary amine. (See: Imines – Properties, Formation, Reactions and Mechanisms)
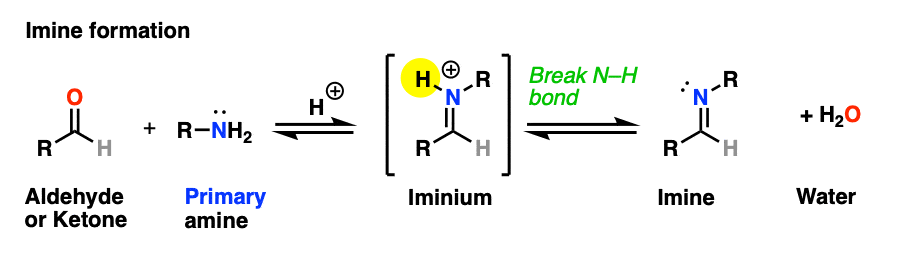
Why only primary amines? Because if a secondary amine is used, the resulting iminium salt can’t be neutralized to an imine through deprotonation of N-H. A C-N bond would have to break, and that’s generally hard to do.
However, if there is a proton on the alpha-carbon (i.e. the aldehyde/ketone is “enolizable”), deprotonation can result in a related species known as an enamine. The formation, properties, and reactions of enamines is the subject of this article.
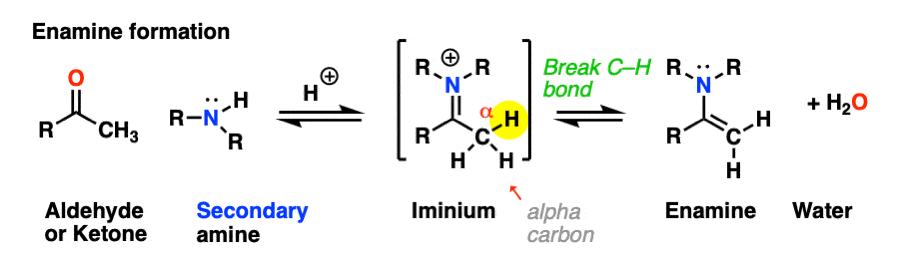
Looking at the bonds that form and break, the major difference between enamine formation and imine formation is that instead of forming a C-N pi bond, we form a new C-C pi bond.
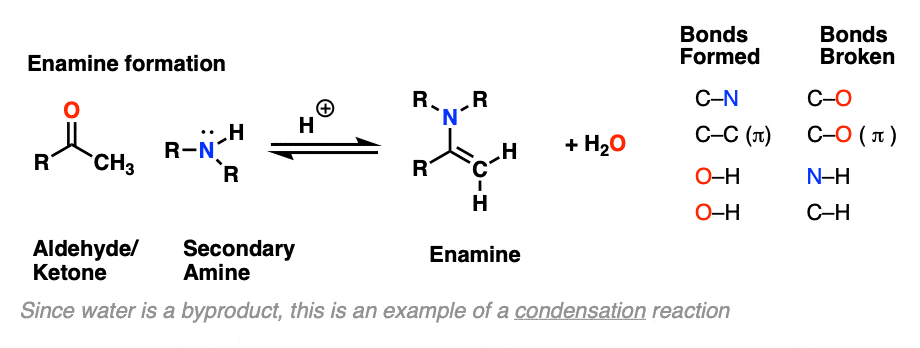
The formation of enamines from the combination of aldehydes or ketones with a secondary amine generally requires an acid catalyst to assist in the loss of water (H2O) from the starting aldehyde/ketone. This is therefore an example of a condensation reaction.
As with imine formation, the reaction is an equilibrium, but removal of water through distillation, or by using a dessicant such as molecular sieves (TiCl4 if you’re hard-core) helps to drive the reaction towards the final enamine product.
Here is a specific example of enamine formation purely for your own enjoyment ( :-) ) (click here for a link to the experimental procedure)
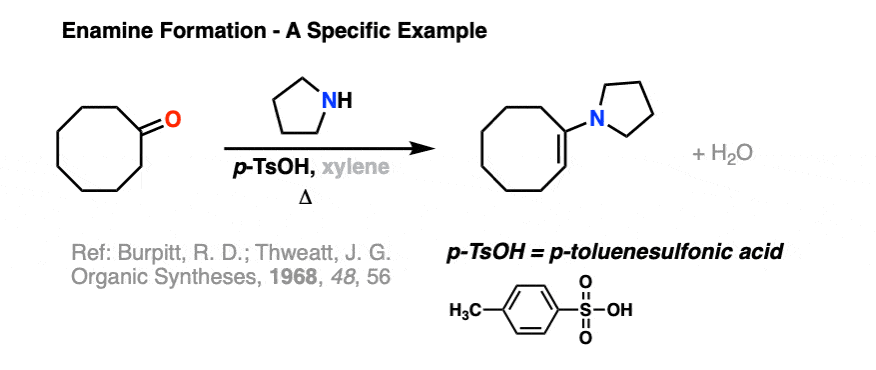
2. The Mechanism for Enamine Formation
In imine formation under acidic conditions, we saw that the mechanism follows the PADPED mnemonic – Protonation Addition Deprotonation Protonation Elimination Deprotonation.
So how does enamine formation work?
Helpfully, formation of enamines also follows PADPED – it’s just important to remember that the Deprotonation occurs on carbon, not nitrogen, and there is formation of a new C-C pi bond. Let’s have a look. [Note 1 for more discussion on the mechanism]
The first step of enamine formation is Protonation of the aldehyde/ketone oxygen, followed by Addition of the secondary amine to the carbonyl carbon. Addition forms a new C-N bond and breaks the C-O pi bond.
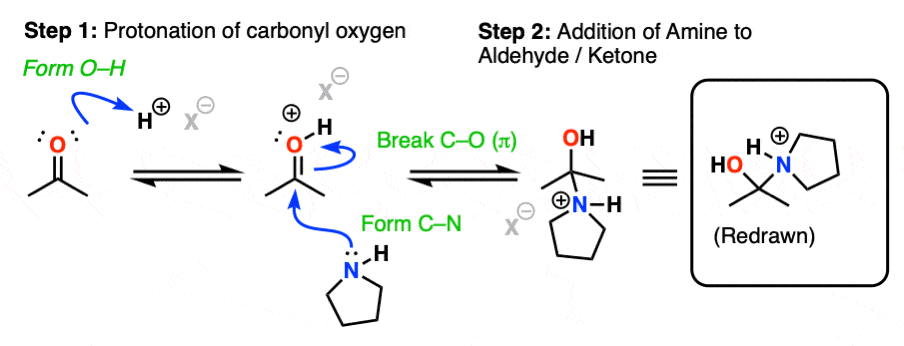
The addition step results in a positively charged nitrogen.
Recall that one of the key roles of acid catalysis is that protonation makes species into better leaving groups (the conjugate acid is always a better leaving group), and if we look at our chart of bonds formed/bonds broken, our leaving group is water. Proton transfer from nitrogen to oxygen is accomplished through Deprotonation of nitrogen with base (e.g. excess amine) followed by Protonation of the OH to give OH2(+).
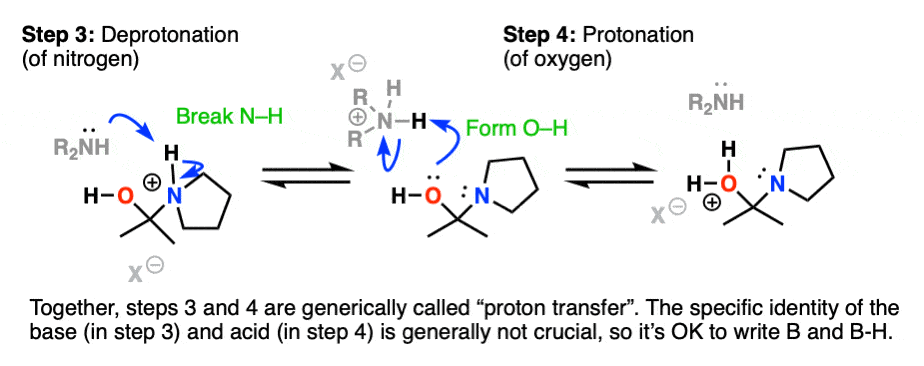
Helpful hint: Avoid showing this as an intramolecular reaction – it’s best to show a base (OK to just draw “B”) acting as a shuttle to pick up and drop off the proton.
Now that there is a free lone pair on nitrogen and a good leaving group (OH2) the stage is set for the reverse of addition, that is Elimination of water to give a new C–N pi bond. This results in an iminium salt. (Note: under mildly acidic conditions this is usually the rate determining step)
As we saw in the previous post, deprotonation at nitrogen is not possible. However, deprotonation at the alpha-carbon (CH3 in this example) can result in formation of a C-C pi bond, and breakage of the C-N pi bond.
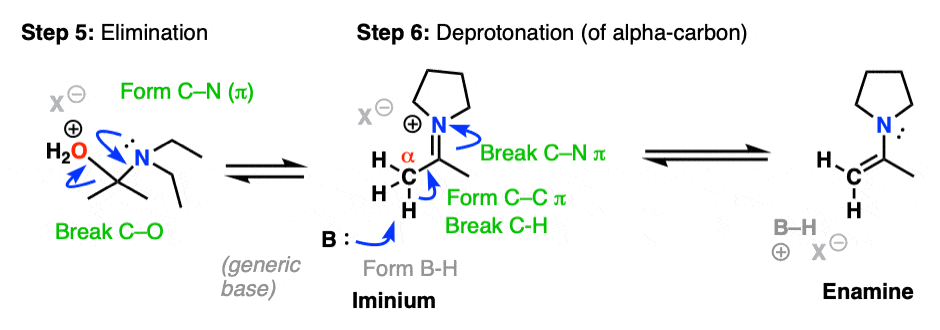
This final deprotonation step gives us our neutral enamine species and a molecule of water.
If you followed along, you’d see the steps were still Protonation Addition Deprotonation Protonation Elimination Deprotonation.
PADPED strikes again! (For 7 more examples, see “Making Music With Mechanisms“)
When an unsymmetrical ketone is used, the tendency is for the less-substituted enamine to be formed (i.e. non-Zaitsev). For more on why this is (good exam question BTW) See Note 2 ]
3. Properties of Enamines (Hint: They’re Nucleophiles)
Now that we have our enamine, you may be prompted to say: “Who cares? It’s just an alkene attached to an amine. What’s so special about that?”
An alkene attached to an amine actually does have some pretty special properties, because that nitrogen lone pair makes enamines capable of all kinds of funky reactions that most alkenes can’t even dream of.
The nitrogen combines with the pi bond of the alkene to give an extended pi system that is capable of resonance.
The nitrogen lone pair of amines is a strong electron donor, and can form a new C-N pi bond with the alkene, resulting in a resonance form with a negative charge on the carbon. Back when we were learning about resonance, we called this property, “pi donation“.
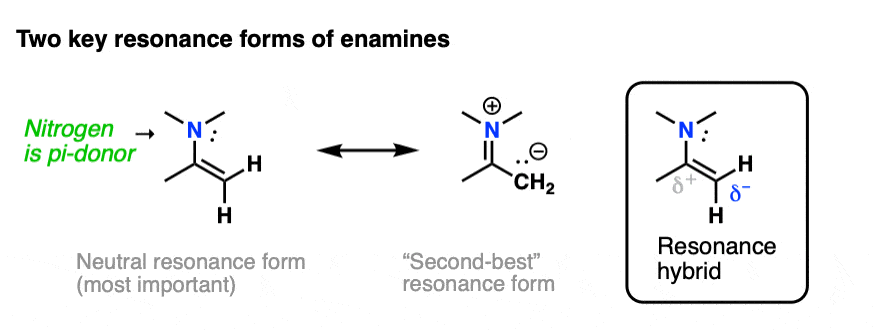
Notice how the carbon has a negative charge in the “second-best” resonance form? That means that carbon in the resonance hybrid will have a significant negative charge density. In other words, it will be highly nucleophilic.
The arrow-pushing is shown in this animated GIF:
You may recall, during the chapter on electrophilic aromatic substitution, that amino groups (-NH2, -NHR, -NR2) are strongly activating ortho-para directors. You may even recall drawing resonance forms where N formed a new pi-bond with the aromatic ring, resulting in a negative charge on the ortho carbon.
hover here for aniline or click this link.
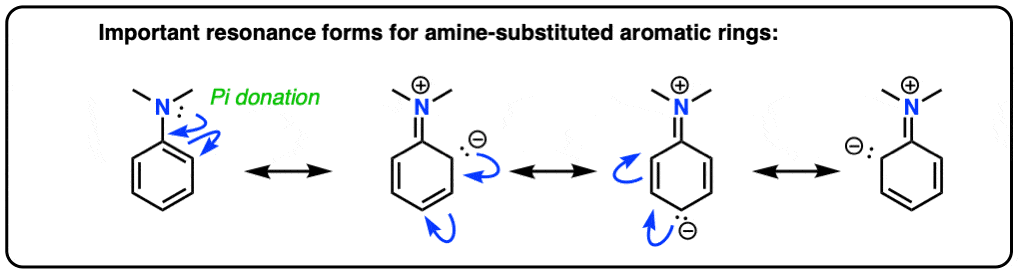{kind=link}
The same phenomenon is in effect here. If you grey out some of the bonds on the aromatic ring and squint, an enamine greatly resembles a strongly activated aromatic ring:
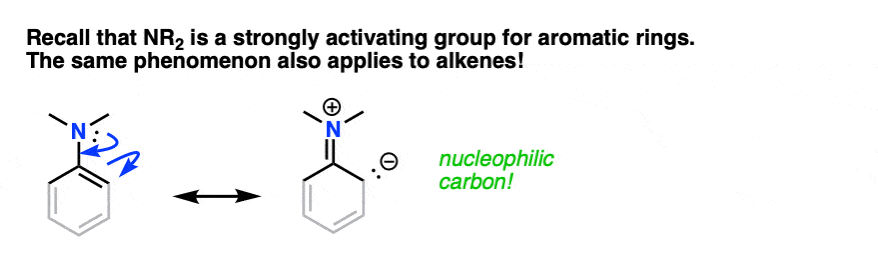
By this analogy we’d similarly expect enamines to be excellent nucleophiles and for them to react with electrophiles.
As we’ll see below, this prediction is accurate!
4. Properties of Enamines – Basicity and Geometry
First of all, a quick quiz.
What do you think the hybridization of the nitrogen in an enamine is?
 Click to Flip
Click to Flip
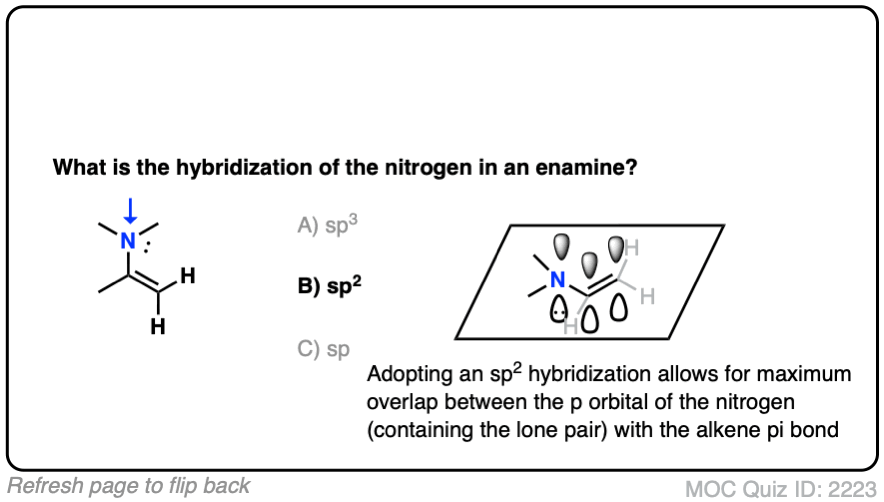
It’s sp2 ! The nitrogen is flat and adopts a trigonal planar geometry, which provides maximum overlap of the p orbital with the p orbitals in the lone pair. Delocalization of the p orbitals more than makes up for the slightly greater steric hindrance encountered in a trigonal planar geometry. [Note 3 – how might we determine this?]
One of the most interesting things about enamines is how basic the alkene carbon is. It turns out that the carbon of enamines is even more basic than the nitrogen of enamines. [Note 4] [Note 5 for more information on protonation of enamines)
This can be observed by treating an enamine with D+ . (Recall that D is the heavy isotope of hydrogen – for our purposes just as reactive, but allows us to “label” molecules.)
A carbon-deuterium bond is readily formed.
What would a mechanism for this reaction look like?
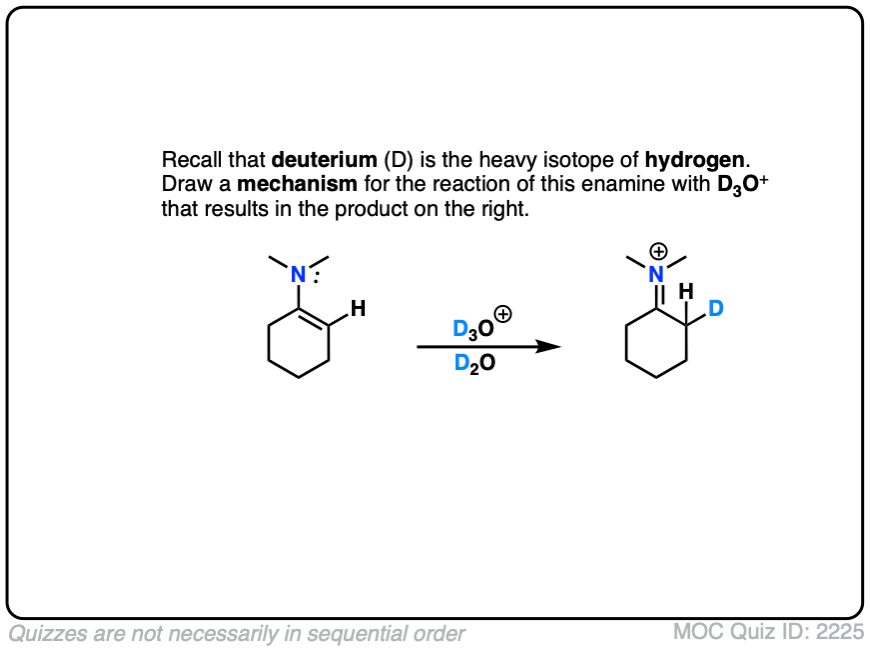 Click to Flip
Click to Flip
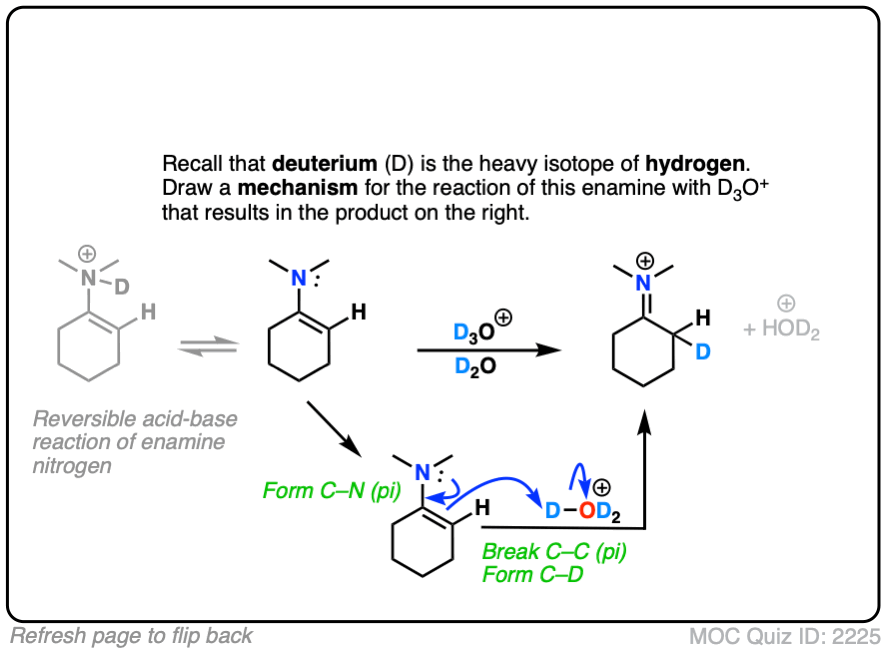
(Note – protonation of the enamine nitrogen also occurs, but it is reversible.)
Note that the mechanism of this reaction is as simple as converting the enamine into the “second best” resonance form, and then connecting that carbon with a proton.
For bonus points – draw a mechanism for the reaction of an enamine with D+ that results in the replacement of H with D.
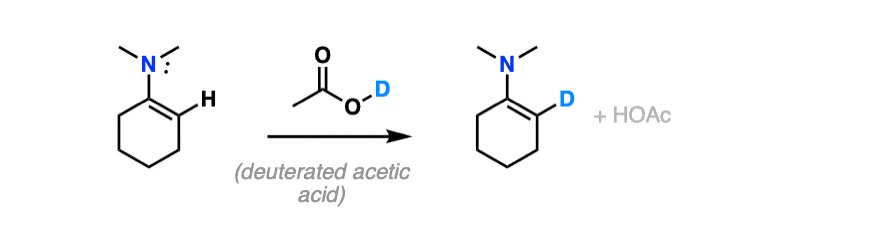
Hint – you might draw inspiration from the mechanism for electrophilic aromatic substitution.
For answer, hover here or click this link.
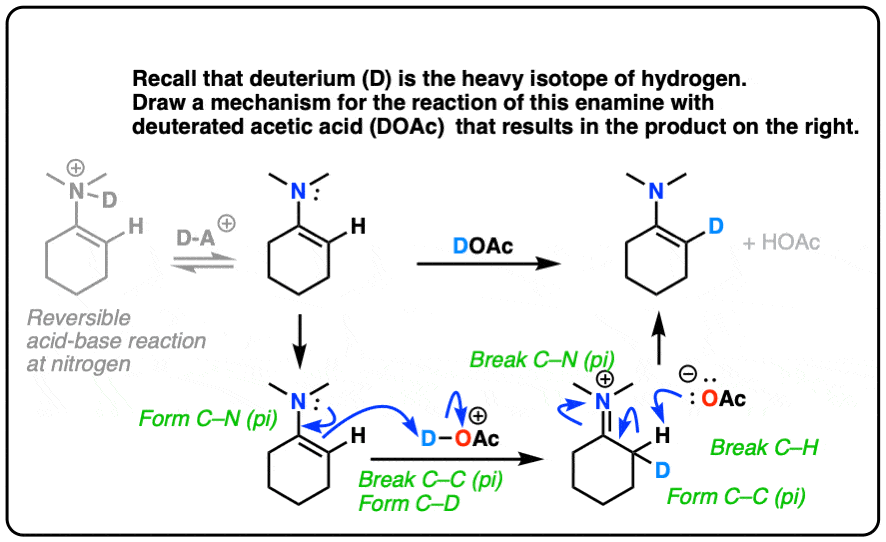{kind=link}
5. Reactions of Enamines: Alkylation
Enamines are significantly more reactive than alkenes towards electrophiles. [Note 6 for some quantification of this]
Alkenes, for example, won’t react with alkyl halides. But enamines will!
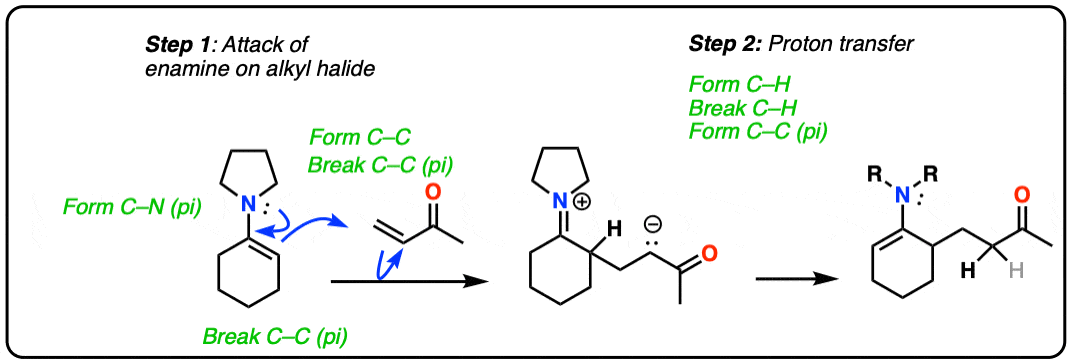
For example, an enamine treated with CH3I will form a new C-C bond. This is an example of enamine alkylation. A new carbon-carbon bond is formed.
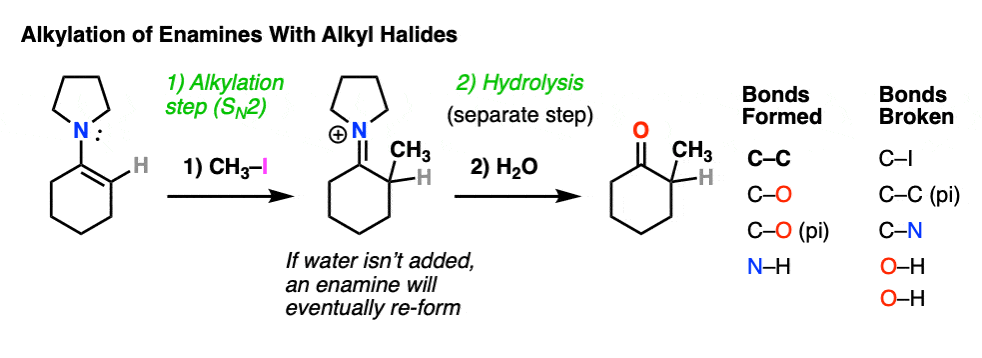
For convenience, water is often added in the workup, so step 2 shows hydrolysis. [Note 7 – in the absence of water, it’s possible for the enamine to re-form]
How does alkylation work? Essentially the same way as protonation, above, except that the enamine carbon attacks carbon as the electrophile instead of H+ (or D+).
This is an SN2 reaction at carbon, with the enamine alpha-carbon as the nucleophile: formation of N-C (pi), breakage of C-C (pi), formation of C-C and breakage of C-X (iodine in this case).
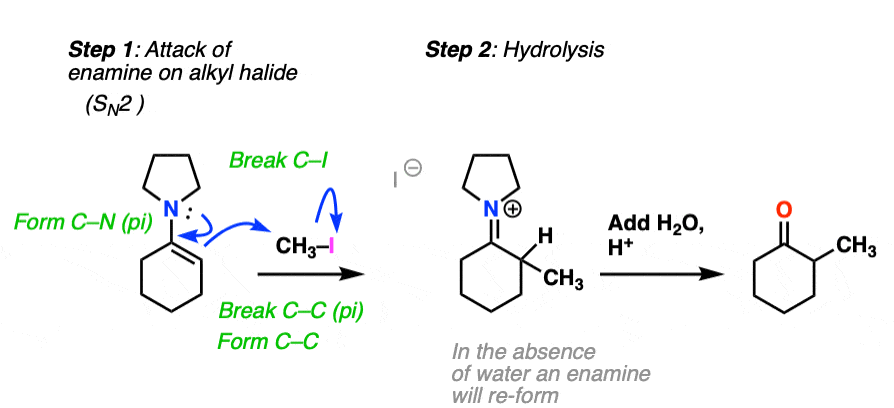
(The mechanism of the hydrolysis step is dealt with a little bit below, click to see it)
For our purposes, you can consider the alkylation of enamines to work reasonably well with most primary alkyl halides. [Note 8 – more detail on this]. It’s a nice alternative to forming C-C bonds through the alkylation of the ketone enolate.
6. Reactions of Enamines: Michael Addition (Conjugate Addition)
Enamines are also nucleophilic enough to perform conjugate additions (“Michael additions”) with alpha, beta unsaturated species such as this ketone below (methyl vinyl ketone).
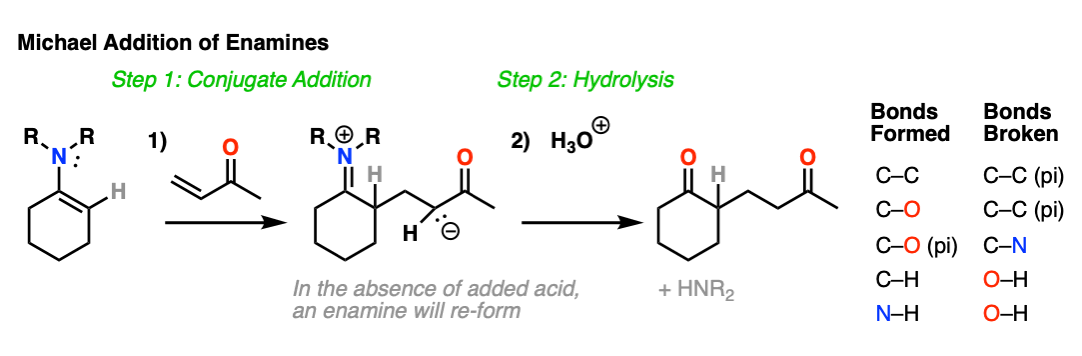
As with alkylation, the usual procedure is to just add aqueous acid after the reaction is complete, resulting in hydrolysis of the enamine and formation of a new ketone.
What’s going on here?
Just as nitrogen lone pairs are pi-donors that make alkenes more nucleophilic, you may recall that C=O bonds are pi-acceptors that make alkenes more electrophilic. A classic example are “alpha, beta unsaturated” ketones (a.k.a. “enones”).
For the reaction between enamines and enones, you might find it helpful to imagine the resonance form of the enamine (negative charge on C) forming a bond with the resonance form of the alpha, beta unsaturated carbon (positive charge on C).
This is not a mechanism – just a rationalization for why the reaction proceeds the way it does. Resonance forms are not equilibria! So be careful not to imply that when you draw out the mechanism.
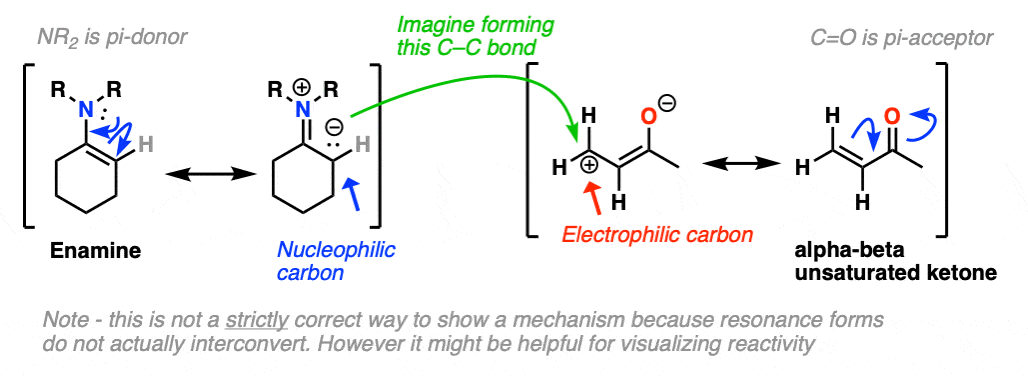
I’ve drawn out the product of the reaction between these two species. hover here or click link. I recommend you start with drawing the ugly version first, and then redraw. The final product is obtained after proton transfer.
{kind=link}
For mechanism, hover here or click this link.
Enamines will also perform aldol reactions with aldehydes, but since this is not covered as much as alkylation or Michael addition and this article is long enough, you can read more about it in this footnote. [Note 9]
7. Reactions of Enamines: Hydrolysis With Aqueous Acid
Enamines are easily transformed back into aldehydes/ketones though hydrolysis with using aqueous acid (H2SO4/H2O is an alternative way of depicting the same thing).
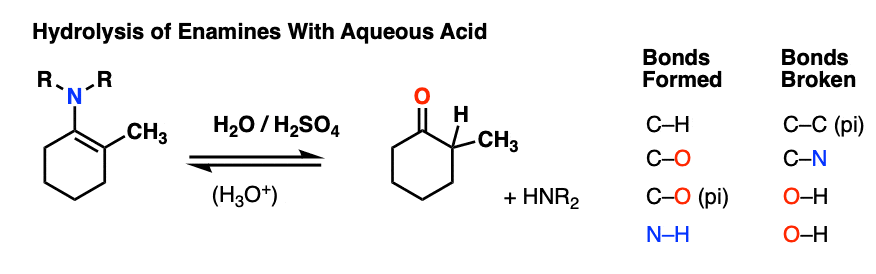
How does this reaction work? Would you believe me if I said, “PADPED” ?
Well, PADPED it is! It’s still an equilibrium, but we’re driving the reaction back towards the starting aldehyde/ketone by using an excess of water.
- Protonation of the enamine at the alpha carbon, followed by
- Addition of water to the resulting iminium ion.
- Deprotonation of oxygen
- Protonation of nitrogen (making it into a better leaving group)
- Elimination of neutral HNR2
- Deprotonation of oxygen to give the neutral carbonyl.
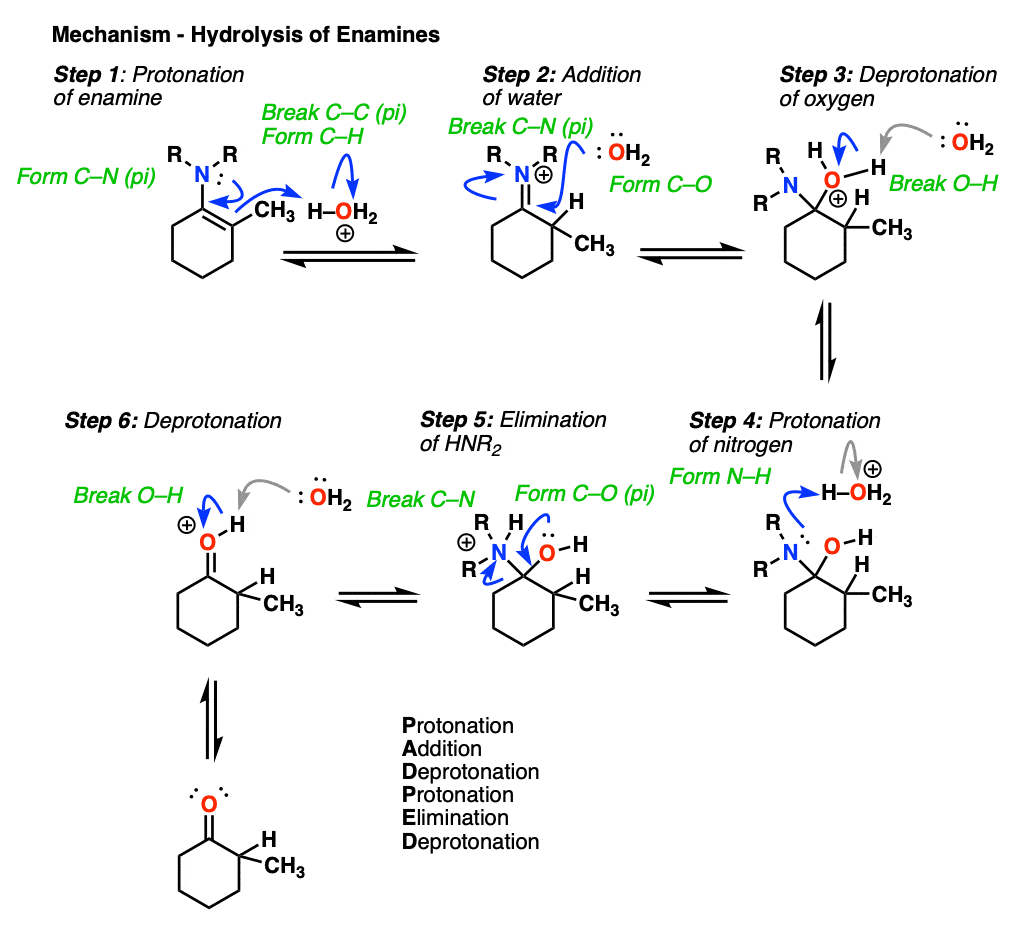
And voilá! We have our aldehyde/ketone back.
8. Summary – Enamine Properties and Reactivity
So what have we learned about enamines? To summarize
- Enamines can be formed through the addition of a secondary amine to an enolizable aldehyde/ketone in the presence of mild acid.
- The mechanism is PADPED.
- The nitrogen of enamines is a powerful pi-donor, making the alkene particularly nucleophilic (just like how amine groups like NH2, NHR, NR2 strongly activate aromatic rings towards electrophilic aromatic substitution)
- Enamines undergo alkylation at carbon with alkyl halides. They can also perform conjugate addition (“Michael reactions”).
- Enamines can be hydrolyzed back to aldehydes/ketones with aqueous acid.
When you think about it, this whole process of ketone –> enamine –> enamine alkylation/conjugate addition –> hydrolysis makes a nice little circle.
We start with a relatively boring aldehyde/ketone. A secondary amine then swoops in, transforming it into an enamine and giving it enhanced nucleophilic powers. This allows the formation of a bond with a desired electrophile. Once complete, hydrolysis removes the (unchanged) secondary amine, and gives us back our aldehyde/ketone, slightly transformed by its adventure.
Kind of sounds a bit like the Hero’s Journey, doesn’t it?
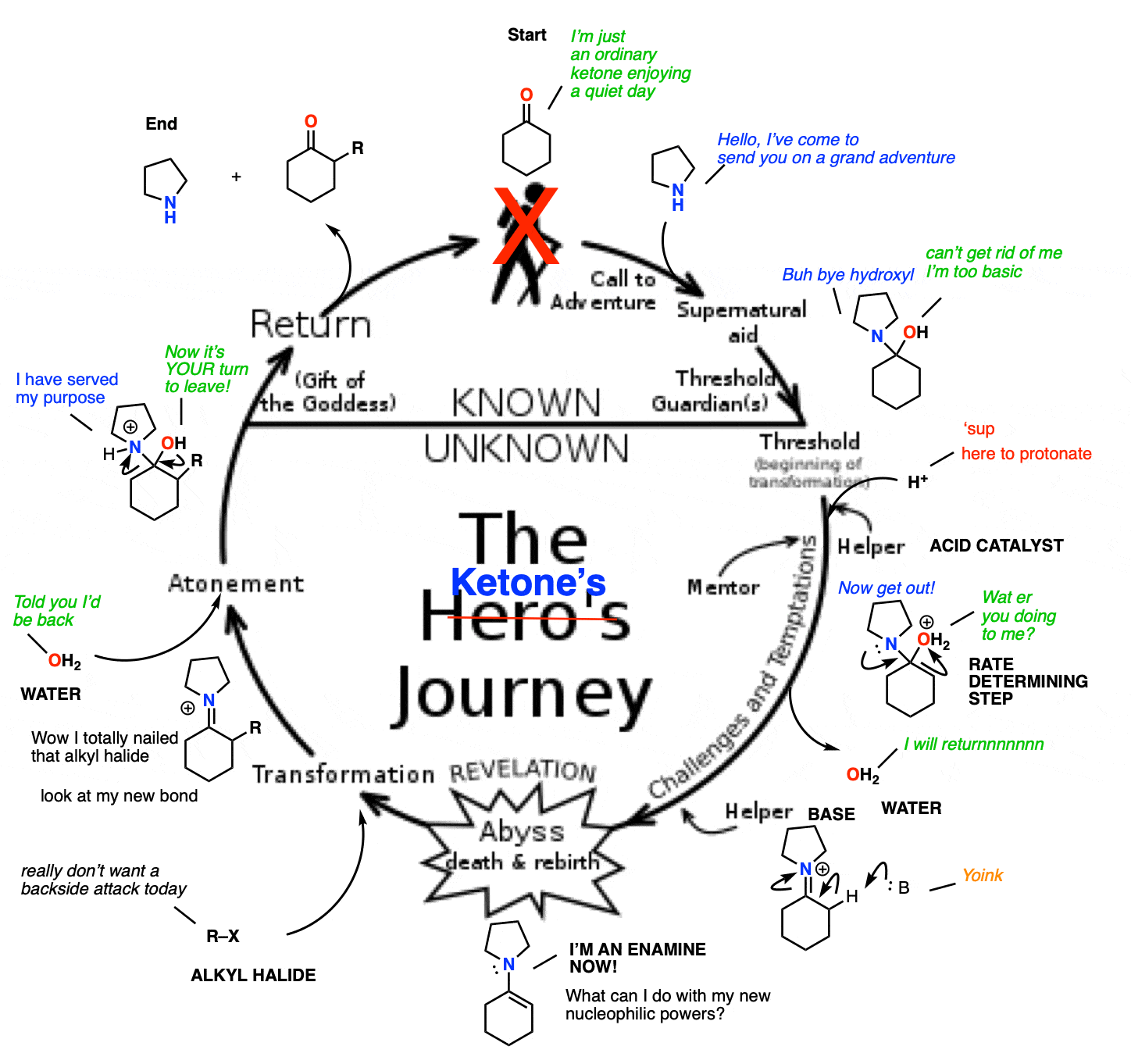
What would be really fancy would be if the secondary amine could be combined with an acid catalyst – then you’d have the potential to turn this into a catalytic cycle.
As it turns out, this has been done [Note 9] (although Nature was there first, by making aldolases).
In fact, the 2021 Nobel Prize in Chemistry was awarded to Benjamin List and David MacMillan for the development of asymmetric organocatalysis. In some of the earliest work (around 2000), List et. al. used L-proline to make a chiral enamine perform aldol reactions, and Macmillan’s group used a chiral amine to make a highly reactive iminium ion undergo electrophilic addition. So the chemistry of these functional groups we learn about in second-semester organic chemistry are still very much an active area of research!
Notes
Reaction guide posts
- Formation of enamines from aldehydes/ketones and secondary amines
- Hydrolysis of enamines
- Alkylation of enamines with alkyl halides
Note 1 – The rate determining step in enamine formation is generally the elimination of water to form the C=N bond. [Ref] It is for this step that acid catalysis is generally required. PADPED is a bit of a pedagogical fib here because acid catalysis is not necessary for secondary amines to add to ketones. For more study on the mechanism, see [this paper] which technically covers hydrolysis but also mentions enamine formation (the two processes obey microscopic reversibility so understanding hydrolysis also helps formation rates).
Also note that the actual acid catalyst is not p-TsOH but ammonium ions formed from the secondary amine and p-TsOH.
Note 2 – Generally speaking, enamine formation favors the less susbstituted double bond. Why might this be?
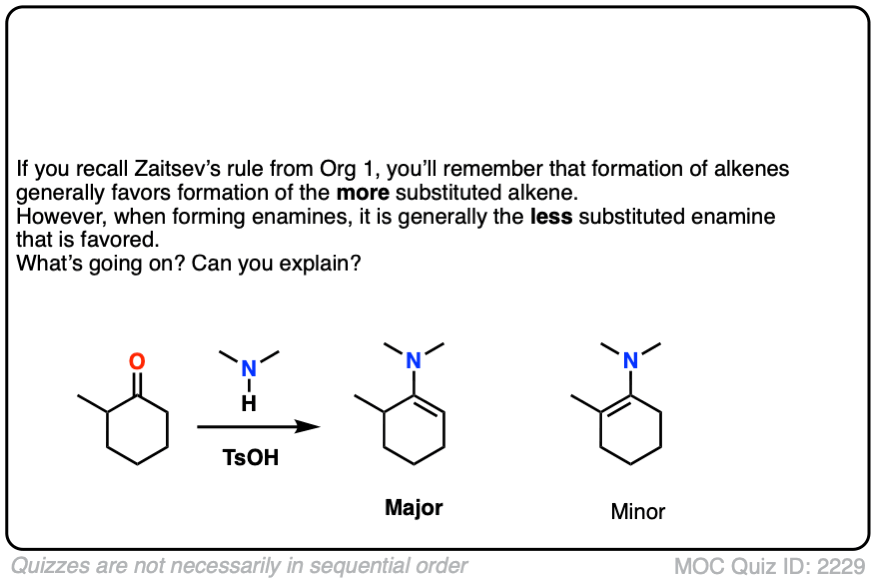 Click to Flip
Click to Flip
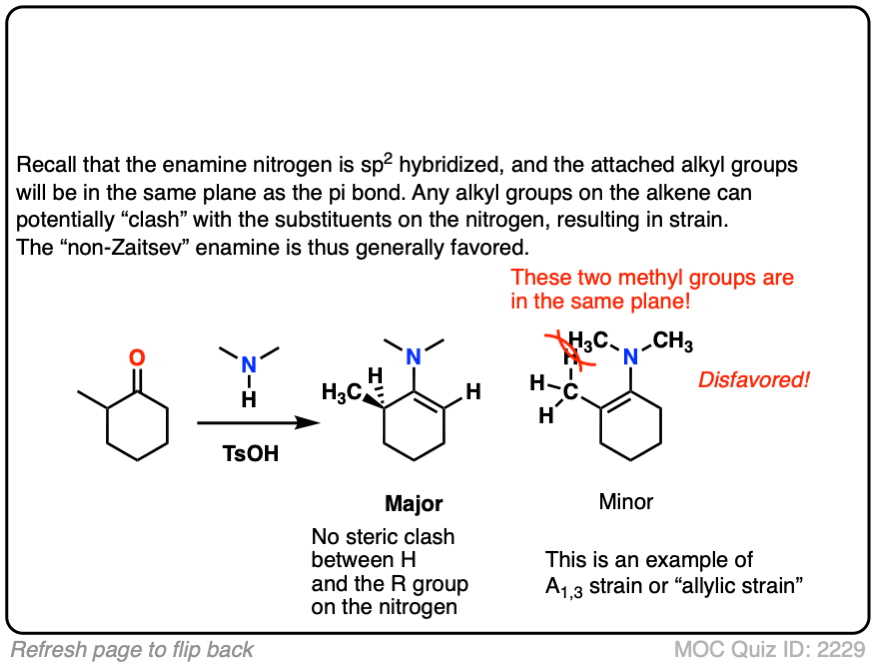
The reason is that the nitrogen is coplanar with the substituent on the enamine beta carbon, which results in them bumping into one another (a phenomenon known as allylic strain.) This strain is avoided when the enamine forms at the less substituted position.
Note 3 – How could we tell that the nitrogen lone pair interacts with the alkene to give an sp2 geometry? There are several important clues.
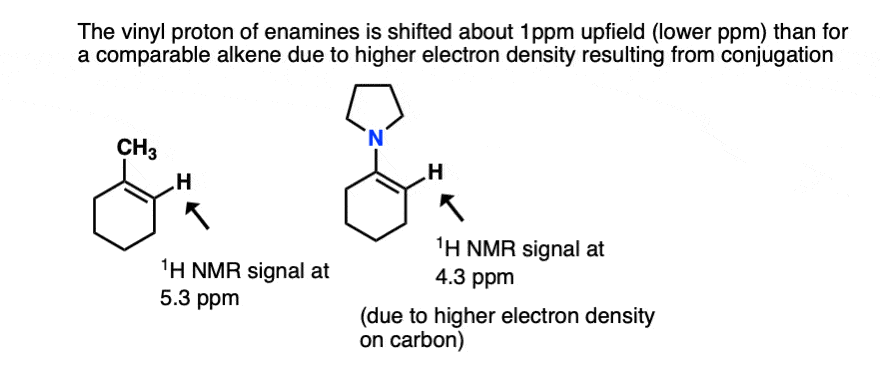
- One is NMR. The typical chemical shift for an alkene proton (“vinyl” proton) is in the range 5-6 ppm (e.g. 1-methylcyclohexene). In enamines this proton is shifted considerably to a lower chemical shift of around 4.5 ppm (the greater the electron density on a carbon bearing a proton, the futher upfield (lower ppm) is the proton chemical shift). In general the lower the chemical shift of the vinyl proton, the greater the overlap.[Ref]
- Another method is the UV-vis spectrum. An enamine will absorb UV light at longer wavelength than a comparable alkene due to conjugation. [e.g. Ref]
- A third is the observation of rotation barriers. If there is overlap between the nitrogen lone pair and the alkene, one would expect to see “partial double bond” character of the C-N bond and therefore the existence of a small rotation barrier. Rotation barriers are observed. [See ref]
- A fourth (dead giveaway) are the many X-ray crystal structures of enamines which unambiguously show an sp2 hybridized nitrogen. [Ref]
Note 4 – What do you think is a stronger base – the nitrogen of enamines or the nitrogen of the corresponding amine?
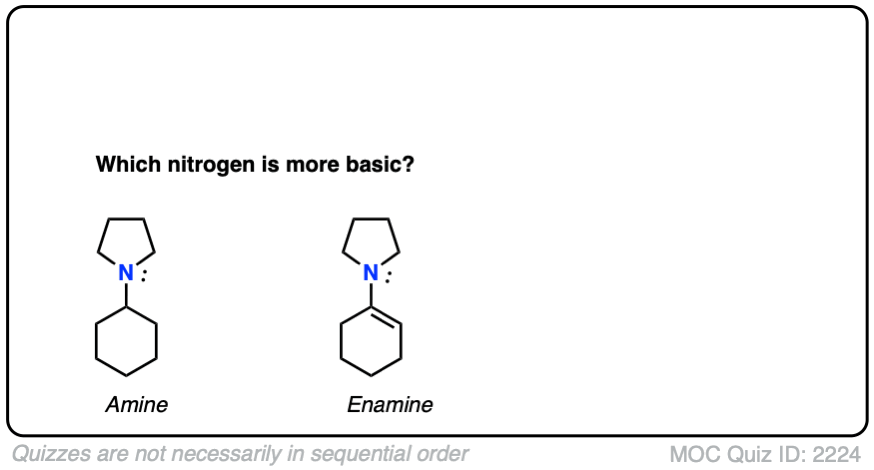 Click to Flip
Click to Flip
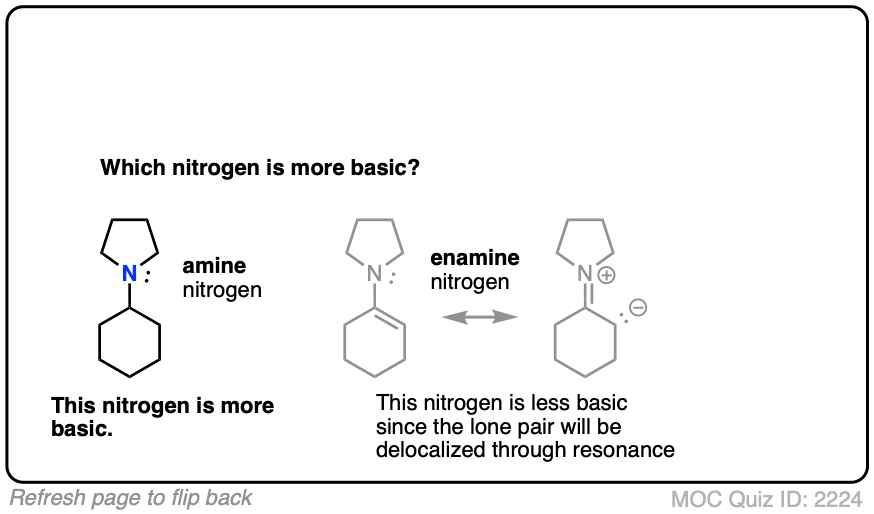
Generally speaking, the nitrogen of an enamine is considerably less basic than the nitrogen of its corresponding amine. Delocalization of the nitrogen lone pair into the pi bond of the alkene results in lower basicity at nitrogen (this is also true for aromatic amines, such as aniline).
Beyond the resonance argument, another way to look at it is that the attached double bond is a better electron withdrawing group than the corresponding alkyl group (recall that sp2-hybridized carbons have more s-character and therefore hold electrons closer to the nucleus, therefore having greater effective electronegativity). Attaching electron-withdrawing groups to amines lowers their basicities.
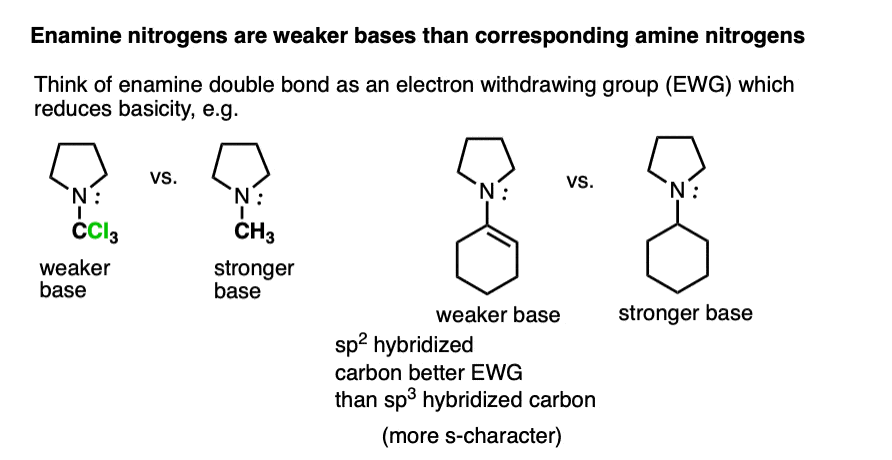
Note that this discussion just applies to the nitrogens. Overall, enamines tend to be more basic than the corresponding amines due to the high basicity of carbon.
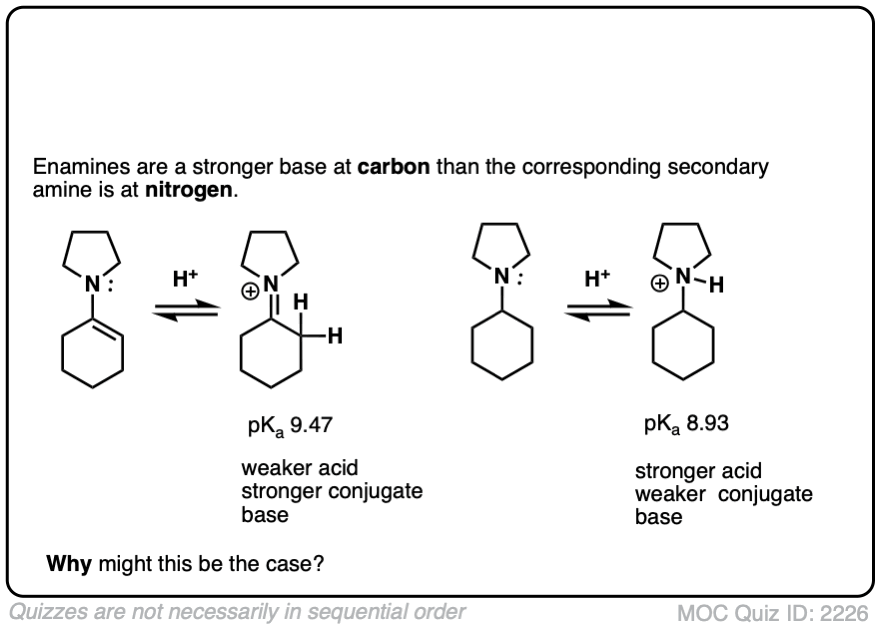
Note 5 – The carbon of an enamine is generally a stronger base than the nitrogen of its amine equivalent. In fact enamines are capable of acid-base reactions with water. [See this ref].
The pKas of enamines have been measured. For example the pKaH of this enamine is 9.47 vs 8.93 for the saturated amine. (pKaH is the pKa of the conjugate acid – recall that the weaker the acid, the stronger the conjugate base, so a higher pKaH means it is a stronger base)
 Click to Flip
Click to Flip
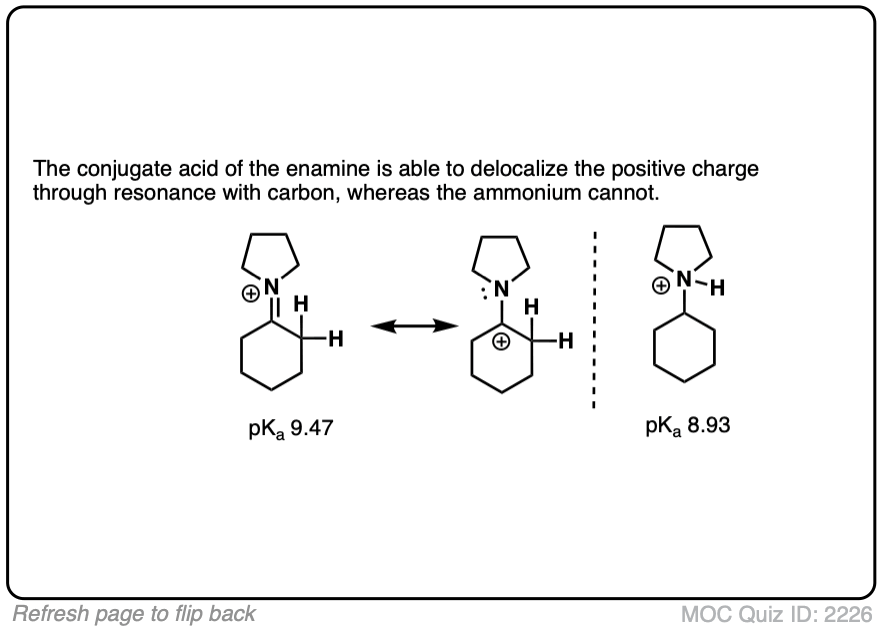
Why might this be? Compare the positive charge. Delocalized between nitrogen and carbon. Versus just a protonated nitrogen.
Note 6 – What do you think is more nucleophilic – an enamine, or an enol ether?
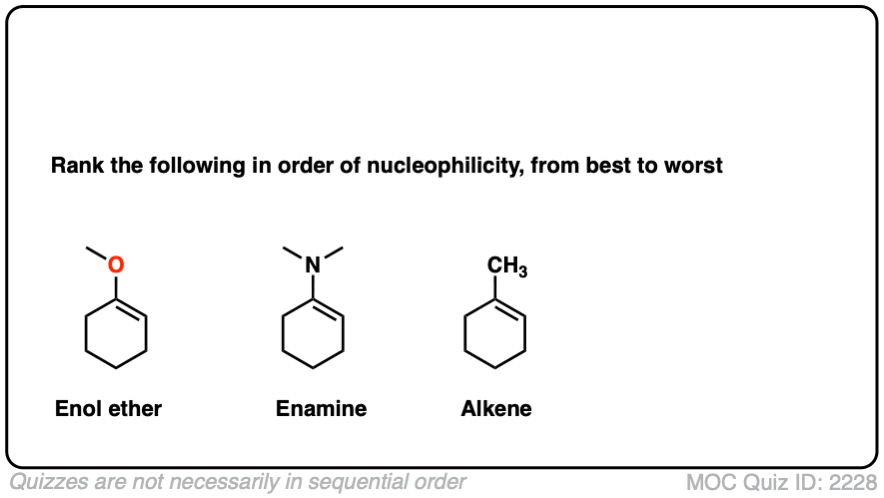 Click to Flip
Click to Flip
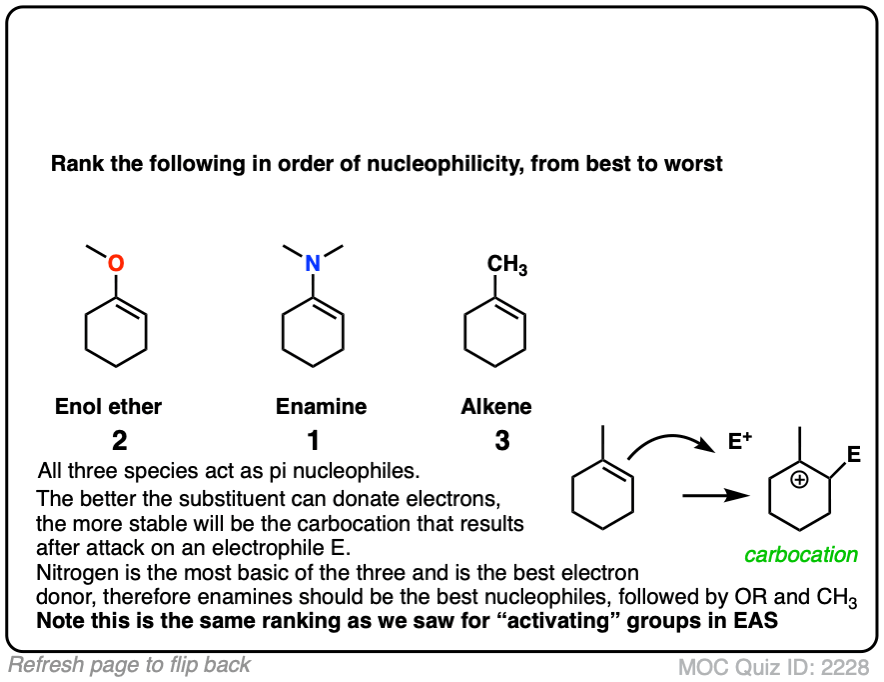
Professor Herbert Mayr of LMU Munich has spent his career developing a scale of reactivity parameters for various nucleophiles and electrophiles. On the Mayr Reactivity Scale, arranged logarithmically like pKa, the pyrrolidine enamine of cyclohexanone is about 15, versus 5 for the silyl enol ether of cyclohexanone and about 0 for 1-methylcyclohexene. This would correspond to a 15 orders of magnitude difference in nucleophilicity between the enamine and the alkyl-substituted alkene!
Note 7 – After alkylation / Michael addition, so long as water is excluded an enamine will be formed from the iminium ion. The new enamine will be the “less substituted” enamine since this minimizes allylic strain (see note 2).
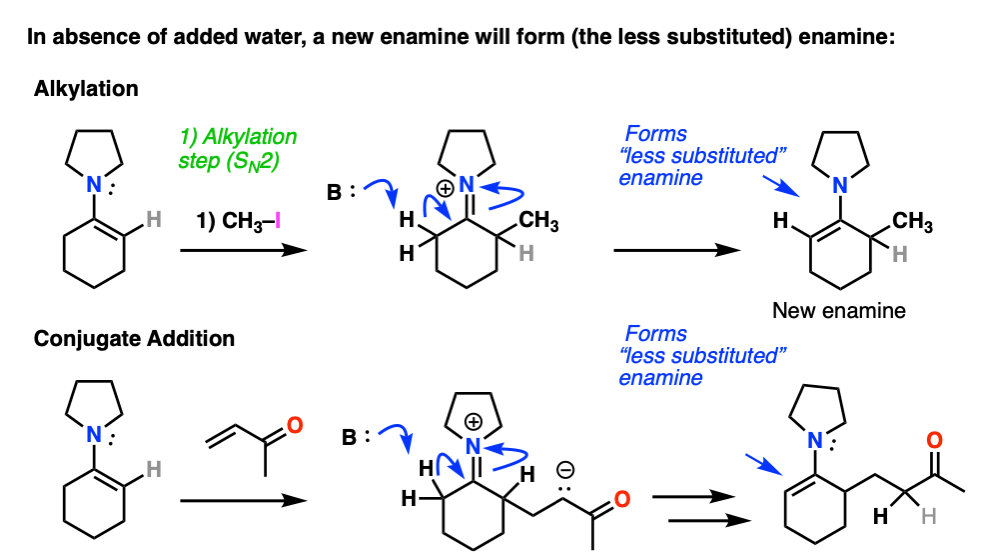
Enamines undergo very easy hydrolysis with water (and mild acid) so in textbooks this “re-formation” step is often skipped and immediate hydrolysis is shown.
Note 8 – In Stork’s 1963 full paper (below) , best yields were obtained with strongly electrophilic alkyl halides such as allyl, benzyl and methyl halides. Simple primary alkyl bromides or iodides were not reactive enough to be practical. One example with butyl iodide gave 44% conversion after 19h with a 57% yield based on recovered starting material; butyl bromide was unreactive.
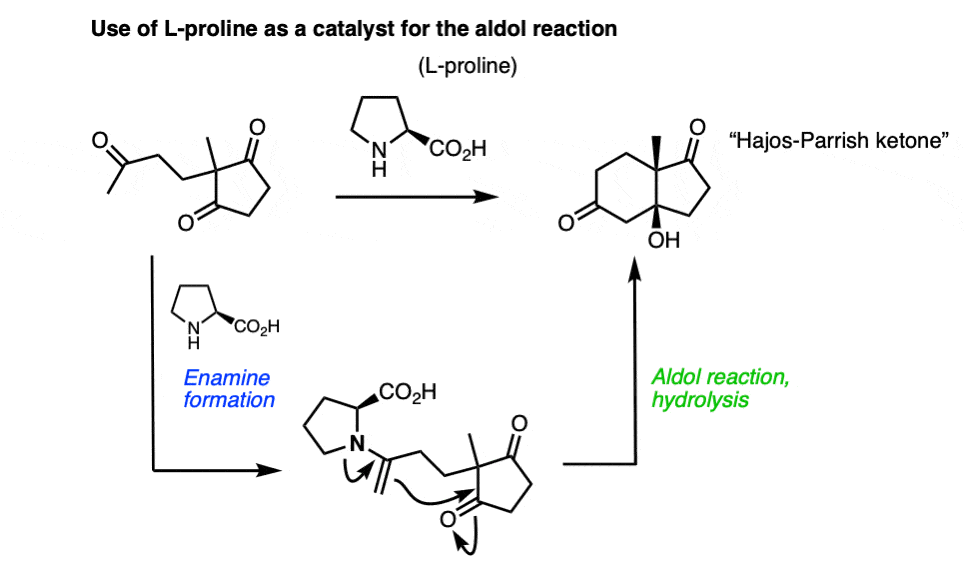
Note 9 – Enamines can also react with aldehydes and ketones to perform aldol reactions. One classic example is the cumbersomely named Hajos-Parrish-Eder-Sauer-Wiechert reaction, which uses L-proline as a catalyst to make an optically enriched enone (the Hajos-Parrish ketone) used in steroid synthesis. The key step occurs via an enamine which performs an intramolecular aldol reaction.
Quiz Yourself!
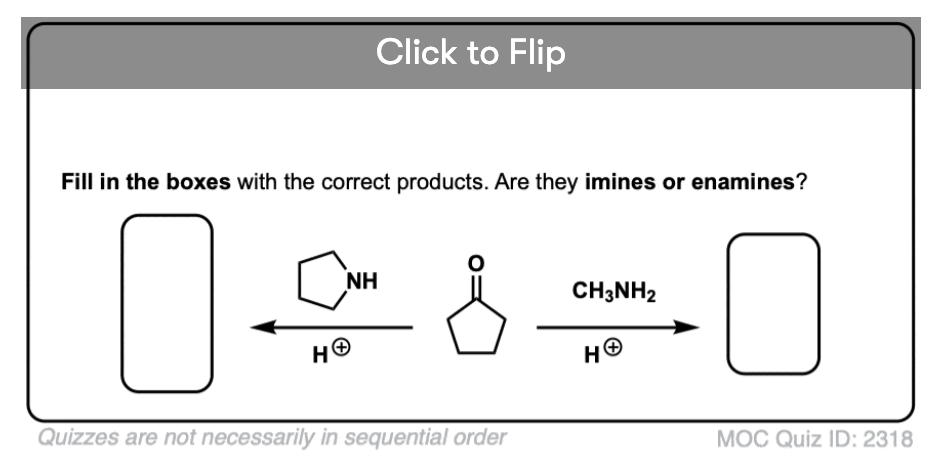
Become a MOC member to see the clickable quiz with answers on the back.
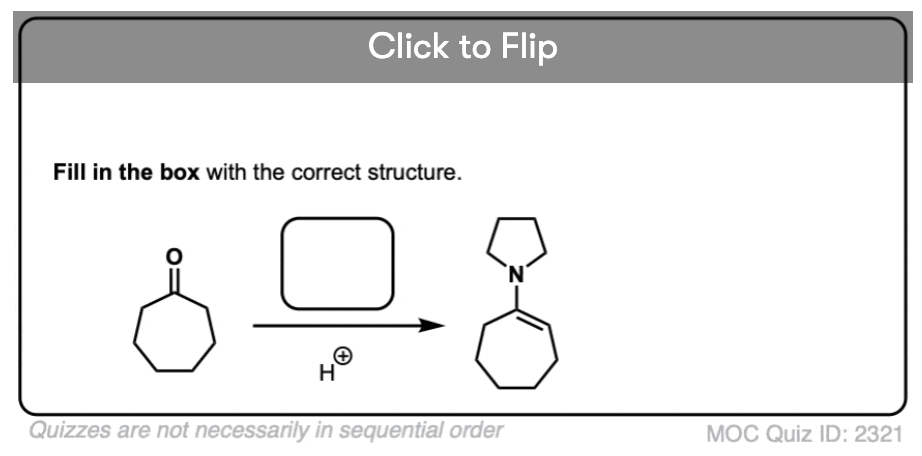
Become a MOC member to see the clickable quiz with answers on the back.
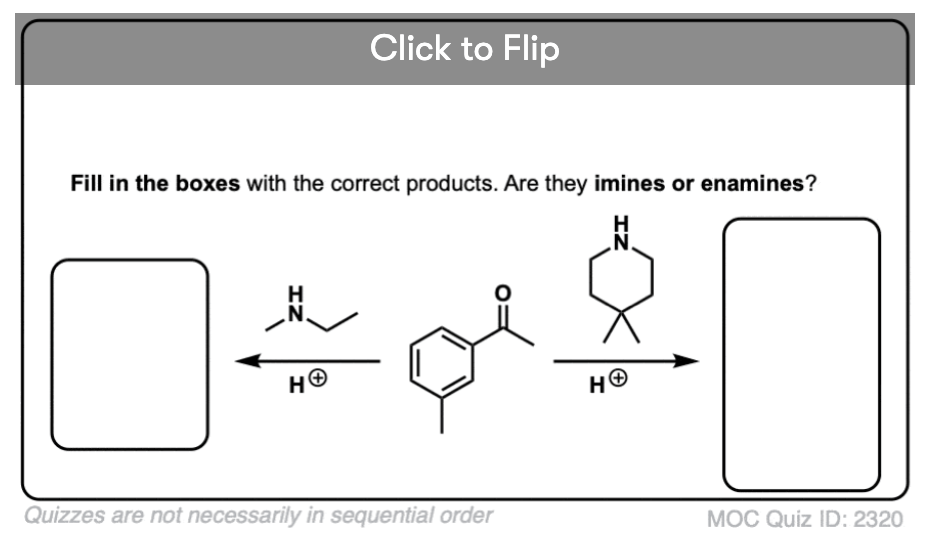
Become a MOC member to see the clickable quiz with answers on the back.
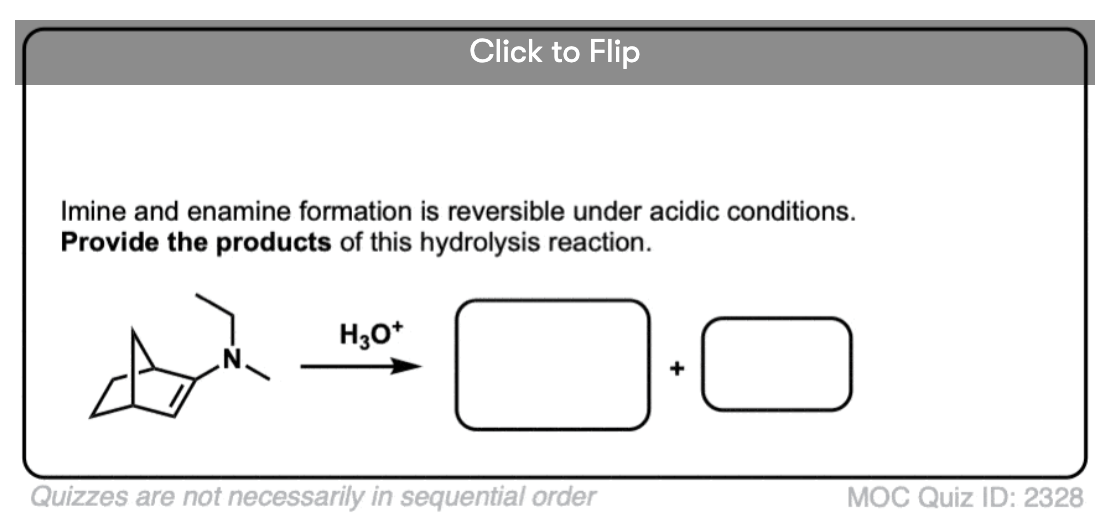
Become a MOC member to see the clickable quiz with answers on the back.
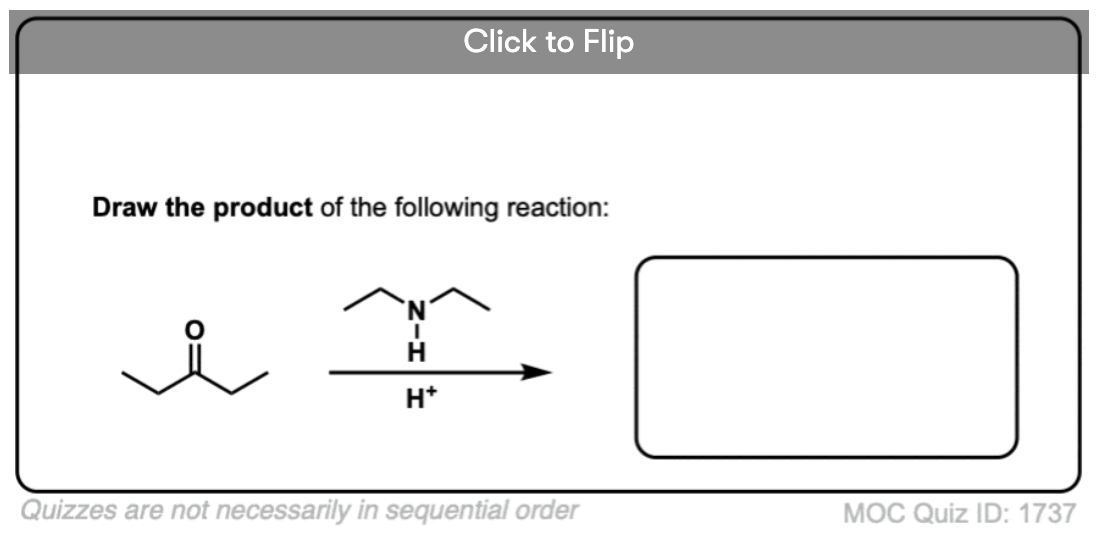
Become a MOC member to see the clickable quiz with answers on the back.
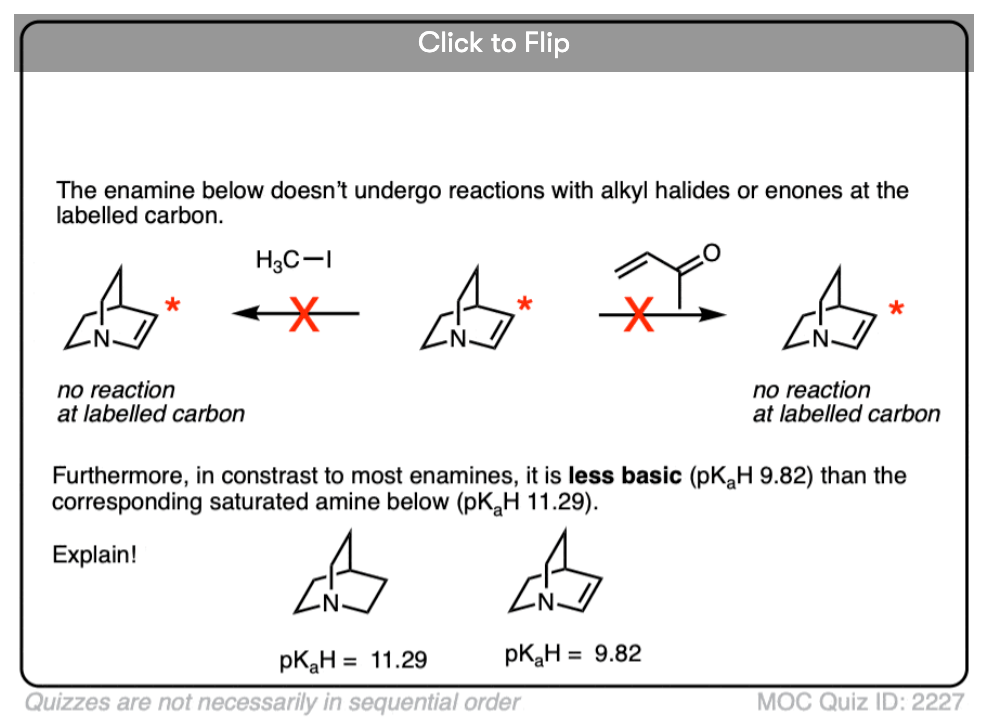
Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading
- Ueber die Einwirkung des Ammoniaks auf Acetessigester
Norman Collie
Just. Liebig. Ann. Chem. 1884, 226 (3), 294-322
DOI: 10.1002/jlac.18842260306
This paper demonstrates that vinylamine reactivity had been known as early as 1884! - Über die Einwirkung von Ammoniak und Ammoniak‐Derivaten auf o‐Acetylaceto‐phenole
Georg Wittig, Hermann Blumenthal
Ber. 1927, 60 (5), 1085-1094
DOI: 10.1002/cber.19270600515
Nobel Laureate Prof. G. Wittig coined the term ‘enamine’ as a nitrogen analog of enol in this paper. - Über einfache Enamine mit tertiär gebundenem Stickstoff
C. Mannich, H. Davidsen
Chem. Ber. 1936 69 (9), 2106-2112
DOI: 10.1002/cber19360690921
Enamine preparation was first made practical by Mannich in this publication - A NEW SYNTHESIS OF 2-ALKYL AND 2-ACYL KETONES
Gilbert Stork, Ross Terrell, and Jacob Szmuszkovicz
Journal of the American Chemical Society 1954 76 (7), 2029-2030
DOI: 1021/ja01636a103
First paper by Prof. Stork on a new method for a-subsitution of ketones via enamines. - The Enamine Alkylation and Acylation of Carbonyl Compounds
G. Stork, A. Brizzolara, H. Landesman, J. Szmuszkovicz, and R. Terrell
Journal of the American Chemical Society 1963 85 (2), 207-222
DOI: 10.1021/ja00885a021
Another classic paper by Prof. Stork on the formation of enamines and their utility in organic synthesis for a-substitution of aldehydes and ketones. Gives examples of Michael additions, alkylations (mostly allyl and benzyl halides) and acylations.
– “Pyrrolidine enamines have been found most generally useful in alkylations with alkyl halides…we have ascribed this to the greater ease of formation of a trigonal carbon ina five-membered ring than in a six-membered one (compare the relative rates of solvolysis of 1-methylcycloalkyl chlorides)
-“Aldehydes can generally be alkylated via their enamines… yields are good only, however, with allyl halides while the simple allyl halides appear to give almost entirely N-quaternary salts”.
– “Simply adding water to an enamine will normally suffice to hydrolyze it to the corresponding carbonyl compound. This is quite unlike the behavior of enol ethers, which are stable in water, and is a reflection of the basicity of enamines toward water… It is evident that all reactions with enamines must be conducted with rigorous exclusion of water”. - CYCLODECANONE
D. Burpitt and J. G. Thweatt
Org. Synth. 1968, 48, 56
DOI: 10.15227/orgsyn.048.0056
The first step in this procedure from Organic Syntheses is an enamine synthesis. - Basicity of Some Mono- and Bicyclic Enamines and Tricyclenamines
A. Gilbert Cook, Michelle L. Absi, and Virginia K. Bowden
J. Org. Chem. 1995, 60, 3169-3171.
DOI: 10.1021/jo00115a037
“A comparison of the basicity of cyclic enamines with corresponding cyclic saturated amines shows that the six-membered ring enamines are only slightly stronger bases than the saturated amines, whereas five-and seven-membered ring enamines are much stronger bases than the saturated amines. This is attributed to a large steric strain present in six-membered enamines”.
Also has measurements of rotation barriers for several enamines. Pyrrolodine enamines have the largest rotation barriers reflecting greater orbital overlap. - Mechanism of Enamine Reactions. IV. The Hydrolysis of Tertiary Enamines in Acidic Medium
W. Maas, M. J. Janssen, E. J. Stamhuis, H. Wynberg.
Journal of Organic Chemistry, 1967, 32, 1111
DOI: 10.1021/jo01279a056
Detailed study of enamine hydrolysis. In weakly acidic solutions the rate of hydrolysis id determined by the rate of addition of water to the C=N bond. - Enamines. II. Factors Determining The Structure of Enamines of 2-Substituted Ketones
W. D. Gurowitz and M. A. Joseph
Journal of Organic Chemistry, 1967, 32, 3289
DOI: 10.1021/jo01286a004
“Enamines of 2-substituted ketones can and do exist as a mixture of the more and less substituted double-bond isomers. The isomer ratio is determined by various steric and electronic factors which affect the overlap between the nitrogen lone pair and the double bond of the enamine. In general the greater the overlap, the greater the proportion of less substituted double bond isomer. In addition, the position of the vinyl proton in the nmr is a reflection of the amount of overlap.” - Comparison of the tautomerization and hydrolysis of some secondary and tertiary enamines
Brian Capon and Zhen Ping Wu
The Journal of Organic Chemistry 1990, 55 (8), 2317-2324
DOI: 1021/jo00295a017
A study examining the stability in solution of various enamines. - Mechanism of Enamine Hydrolysis
Paula Y. Sollengerber and R. Bruce Martin
J. Am. Chem. Soc. 1970, 92, 4261-4270
DOI: 10.1021/ja00717a021
“Rates of hydrolysis of the morpholine, dimethylamine, piperidine, and pyrrolidine enamines of propiophenone [were] investigated in the pH range –3 to 14 at 25°…. In weakly acidic solutions (pH 1–6) carbinolamine formation via uncatalyzed attack of water is rate limiting…. In neutral and basic solutions the rate-limiting step is either [protonation of the alpha carbon or attack of water/hydroxide ion]. ”
“From observations on synthesis rates from cyclic ketones and other considerations it appears likely that carbinolamine dehydration is the rate-limiting step in the synthesis of many enamines”.
How can we test between enamines and imines, enamine and allylamine?
Why don’t alkenes react with alkyl halides. Is it to do with the orbital energies? Thank you.
They just aren’t very good nucleophiles. A carbocation would have to form, which is generally disfavored unless a Lewis acid is used such as AlCl3 to “pull” the halide off.
James,
You stated that an oxime has the C=N double bond with two organic constituents on the carbon and a hydroxyl group on the nitrogen. If the hydroxyl group were instead an ether, would it still be an oxime or would it have a different name?
I’m curious as I found a pharmaceutical compound (Roxithromycin) that wikipedia says contains an N-oxime side chain. However, every source I’ve seen on oximes says they have an R1R2C=NOH general formula, not an R1R2C=NOR3 formula.
Thanks,
William
Hi William – it would be an oxime ether if there is an R group on the oxygen. Do a search for “oxime ether” and you should find lots of information.
Hello James.
How stable aliphatic imines are if the water is absorbed by a sulfate? It’s often said they polymerize, but that’s it. No more information.
Where is it said that they polymerize? Using MgSO4 or Na2SO4 to make an imine is pretty standard practice.
Thank you for your very informative website, I often consult it when I am in doubt about organic reactions.
This reaction mechanism has puzzled me over the last few days. The pKa for deprotonating a ketone is around -7, whereas many protonated amines of interest are around 10. If we only reason in terms of protonated ketone and amine and unprotonated ones, then we see directly that in the pH range 3 to 7 the product [protonated ketone][unprotonated base] should remain rather constant, and be extremely low.
After doing some digging, I find that some authors prefer writing a concerted mechanism, and some attack an unprotonated ketone. The latter is certainly less electrophilic. However, at pH 5, it is 10^12 times more abundant, which in free energy terms means RT ln (10) =69 kJ/mol=16.5 kcal/mol extra in favor of this pathway.
If we opt for the latter mechanism, we should expect the rate to depend on pH, but for a different reason: at low pH, there is not enough amine to attack ketone (protonated or not). At high pH, the elimination of water becomes rate-limiting (since the H2O+ moiety will spend more time as an OH moiety).
Of course, my thinking is based on pKa values in water, maybe the mechanism is quite different in organic solvents. What are your thoughts on this?
Hi Alex, you have a great point, recently brought up in the comments of this post. https://www.masterorganicchemistry.com/2017/09/01/reductive-amination/
I think EB is correct in saying that the first step should be addition of the amine to the carbonyl, and that the proton source assists with departure of the resulting OH. I’m going to put fixing this in my correction queue.
Correction: imines formed from ketones are referred to as ketimines. Ketamine is a drug with ketone and amine functional groups.
Thanks for catching that typo, Matt.
What will be the product if there is an alkyl group attached to the cyclohexanone at the alpha position which is then made to react with the secondary amine?
Hello sir,
Why are imines considered less stable than amines and how can we orderize different imines based on stability like conjugated, non conjugated, present with cyclic products,or benzene. Plz help
They hydrolyze relatively easily in the presence of water.
Question about an example that lead me to this page involving reaction of keytone with primary amine forming an imine intermediate. You said sodium cyano BH3 was mild enough the reduction could be carried out in the same vessel in one step. I can see how that will work, but what kind of yield is achieveable? Removing the imine product should shift eq. to favor more product formation which is great, but what happens as the reduced second product concentration begins to rise? Would you not risk the desired final product only being a secondary amine bumping into the keytone (I assume the keytone is solvent and primary amine con. used to control rate and yield) and reacting again to lead to an unwanted enamine side product? Would steric hinderance have any affect on preventing this?
Unless thats not an issue, seems to me a better yield could be achieved with little to no side products by isolating the imine and reducing in a second step. Or maybe using the primary amine as solvent and lower con. of keytone so theoretically “all” keytone would be used, then product could be separated from solvent and maybe more keytone then added for further production?
Also, the thermodynamics…this should occur at a decent rate im guessing at 20-30C with some time and gentle mixing? Heat could increase rate, but may also destabolize reducing agent to the point of attacking the keytone? And last, since imine removal drives first reaction toward making more imines, at what point would water build up become a problem? Would it still need removed? Too much h2o possibly start altering initial pH or cause rxn to slow or stop due to solvent interactions through hydrogen bonding on the small primary amine?
Hello,
Do you have any examples of Beta carotene synthesis that involves imine formation? I received it on an exam but cannot find anything similar
James,
I am 75 years old. Biochemist. Still full time employed. Have developed a lot of analytical methods for industrial hygiene atmospheric pollutants (e.g., NIOSH method 9109). Still doing so. Your web site has been extremely helpful. We have LC-MSs and GC-MSs. But the LC-MSs are always so back-logged with work to do, I try to find ways to derivatize analytes to make them GC-able. Current challenge is Paraquat. Your web site discussions on enamines, imines, sodium borohydride, etc. help me understand what is going on in the process of turning pyridinium rings of Paraquat into tertiary amines. I read literature on how others have done this, but it is very satisfying to understand what the mechanisms are behind reported procedures in the literature. It also helps me to figure out if there are ways the procedures can be improved. Thanks again. John M. Reynolds
Dear James,
I am trying to react an aldehyde with ammonium chloride. At a low pH, the majority of the species are ammonium and not ammonia. However, at a higher pH the reaction will proceed slowly, as you mentioned. Do you have a recommendation of the optimum pH required for the imine formation?
Best pH is around 4 or 5 .
Hi! I was wondering if imine formation can happen without an acid catalyst? Thanks.
Not required, but it helps.
Ah! This makes so much sense!
Might I suggest the Learn 6 reactions for the price of one article be linked?
Thanks so much! Doc!
Dear James,
Thank you for your informative website! My question is with regards to imines formed in the absence of an acid catalyst. I’m assuming that the protons that eventually leave with the oxygen as water by product come from the amine group. If so, it is correct to draw the formation of the intermediate formed with an arrow showing the N-H bond disconnecting and the movement of the H to the oxygen to form OH? I’m trying to draw up the mechanism but I’m not sure if I’ve done it right. Thanks!
What you’re describing is a process called “proton transfer” where the protons on nitrogen ultimately are removed and protons are added to oxygen. The exact mechanism instructors use can differ considerably. In the short way, which you describe, an arrow is drawn that shows the oxygen deprotonating the nitrogen directly. A more painfully long way to draw it (but ultimately more correct) is to invoke some kind of exterior base (like water) to deprotonate the nitrogen, and then (in a second step) to act as an acid, protonating the oxygen.
It all ends up being the same thing.
https://www.masterorganicchemistry.com/2010/04/30/the-magic-wand-of-proton-transfer/
Dear James,
wouldn’t acid first protonate amino group and not ketone/aldehyde? Amino group is weak base thus it should accept proton easier than C=O? Yet in this reaction oxygen is the one accepting proton. Why is that?
Thank you
Yes, it protonates the amine but not irreversibly. Remember if the amine was protonated irreversibly there would be no nucleophile to attack the carbonyl carbon. That’s why pH is essential. If it’s too acidic, the amine is protonated irreversibly, if it’s not acidic enough (pH 7 or higher) then the reaction is slow. The “sweet spot” is about pH 4 or 5.
Dear James,
Thank you for a very useful website which I refer to regularly. Please could I ask whether the less hindered substituted alkene would be formed in the last part of your diagram for Alkylation of Enamines (Stork Enamine Reaction) because of allylic strain?
Thanks again and best regards, Ian
Yes – exactly! Allylic strain between the alkene substituent and the nitrogen substituent.
Hi! I am doing reductive amination in which imine is a intermediate. I am using aliphatic aldehyde and aliphatic amine in presence of Ra-Ni. CAn you suggest me what is the suitable water scavenging agent for the reaction?
I don’t believe scavenging water is necessary since once the imine is reduced, it will be stable and will not reverse in the presence of water.
Hi,
In your note you state that the acid is not an absolute requirement for the reaction.. It means that the reaction can still occur? even in standard temperature?
thanks
Yes, especially with aldehydes, acid is not an absolute requirement.
What would be the resulting product with H2S gas reacting with imines? When tried to to bubble H2S gas on organic imines it resulted to a solid formation. What is the component of this solid? Why it does formed?
I like this website! I’m studing a sintesis of the Penicilin V and found a obstacle, but this page help me more than a book. thanks!
Looking for the distinguishing factor of a enamine, three bonds and a lone pair? but more important: this rules. your enthusiasm rules. I love chemistry too.
Enamine: alkene directly attached to an amine. Part alkene, part amine, kind of like a spork is part spoon, part fork. Thank you for the last comment too
Hey James, I have a question about the reaction of imines. I can’t seem to find anything online about them except for their formation!
Do you know what the outcome is when you react an imine with an acyl chloride? I can’t work out if the nitrogen attacks the C=O carbon or the oxygen attacks the C=N carbon.
Thanks.
When you treat an imine with an acyl chloride, the nitrogen attacks the carbonyl carbon. (Nitrogen is more nucleophilic than oxygen). This forms an N-acyl imine (or iminium, depending on structure) which is a great electrophile. Kinda like in this example (far near the bottom) – http://en.wikipedia.org/wiki/Pictet–Spengler_reaction
That’s a great help. Thank you!
Hello James can you please throw some light on major product formed when a 2-methyl cyclohexanone reacts CH3I via enamine ? Also Explain which type of enamine formation dominate ie kinetic or thermodynamic enamine ?
When you draw out the enamine that would form on the “most substituted” side, you’ll see that the methyl group on the 2-position has a steric clash with one of the alkyl groups on the nitrogen. It’s called “allylic strain”. Enamines tend to form on the less-substituted “alpha carbon”.
I have a question. How to separate aromatic imine from the reaction mixture? Thanks!
Depends on your reaction mixture. Some aromatic imines are quite crystalline. The problem is that they are readily hydrolyzed. I would suggest going to chemicalforums and giving out your specific reaction conditions – there’s a lot of helpful people there (myself included) who can assist with this type of practical question.
how to remove water in the imine formation reactions?
there are different methods which does not involve water fromation like dehydro halogentaion, azawittig reaction. it depends on the type of imine you are working on.
True. This post is written for people taking organic chemistry for the first time, who probably don’t come across the aza-Wittig reaction and other alternative methods of making imines.
please give one simple method for the synthesis of imine and how can we remove the water formed in the reaction?
Depends on the type of imine, and the reactivity of the amine and aldehyde involved. For simple imines with electron rich amines and aldehydes, anhydrous magnesium sulfate [ Mg(SO4)2 ] is usually sufficient. One can also use other dessicants such as silica gel or powdered 3A or 4A molecular sieves. For stubborn imines, heating in the presence of catalytic acid with an affixed Dean Stark trap using toluene or benzene as solvent will do the trick. Hope this helps – James