Fun and Miscellaneous
The Organic Chemistry Behind “The Pill”
Last updated: September 24th, 2022 |
The Most Interesting Man In The World passed away the other week.

No, not THAT guy. He’s just an actor. The OTHER guy.
The OTHER guy is Carl Djerassi, organic chemist, professor, author, playwright, and most notably, “Father of the Pill”.
Lots of people these days – especially startups – talk about changing the world. Unlike, say, Udacity, the invention which Djerassi had a major hand in actually did change the world. It’s estimated that over a third of women in the USA have taken some type of birth control pill during their reproductive lifetime, for example, and over 100 million women worldwide are taking it now. By giving women control over their own reproductive process, the technological advance laid the groundwork for the social change of the 1960s.
Today we’re going to take a tour of the organic chemistry that went into the synthesis of the Pill’s original key ingredient and show that the 6 steps involved are entirely comprehensible to an involved student of organic chemistry who is about halfway through Org 2.
A Little Background
It had been known since the 1930s that large doses of steroid hormones could inhibit ovulation in mammals. Explaining exactly how this works is best described elsewhere – check out this beautiful infographic by Compound Interest – but the basic idea is that high concentrations of sex hormones (such as progesterone, below) fool the body into thinking it is pregnant [BTW this drawing is from my dad’s medical school notes. Thanks Dad.]
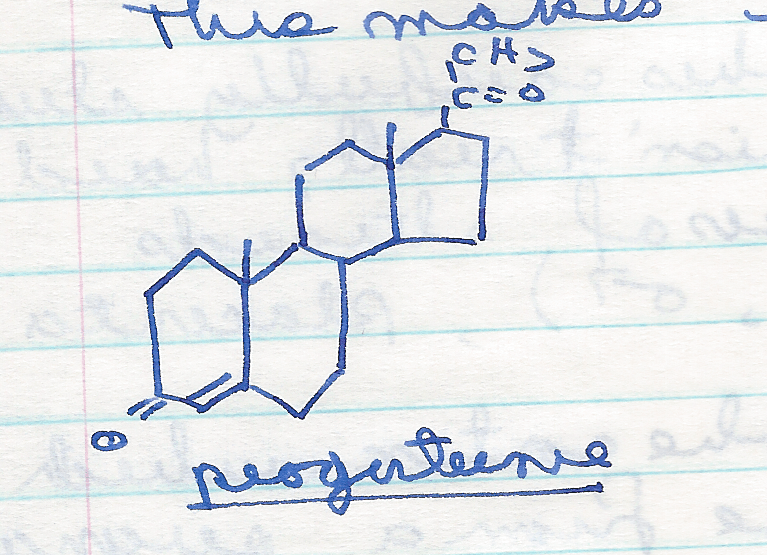
So if this was known by the 1930’s, you might wonder why the 1940’s or 1950’s weren’t the decade of the sexual revolution, with women self-administering progesterone and throwing off the shackles of conventional sexual mores?
Two problems:
First, in the 1930s progesterone was extremely expensive – at over $80/gram, far more precious than gold. This is because it had to be isolated from animal ovaries and the process was extremely inefficient (to say the least).
Secondly, progesterone is rapidly metabolized in the liver, meaning it was not practical for use in a pill. In the late 1930s there was some initial work on developing chemical relatives of progesterone [“progestins”] that mimicked the effects of progesterone but were more biologically active when taken orally, but the difficulty of obtaining large quantities of steroid starting materials was a huge limiting factor.
Sex Hormones – The Mexican High Tech Industry Of The 1940s
The progesterone supply problem – which is well worth a blog post on its own – was solved in the late 1930s by American chemist Russell Marker, who developed a method to produce progesterone and other sex hormones from steroids present in the Mexican yam. Notably, in the first batch process, 10 tons of yams yielded over 3 kg of progesterone, with a value of over $3 million in today’s dollars. Marker moved to Mexico to be closer to the source – one presumes that shipping dozens of tons of Mexican yams across the border to the USA was problematic – and hired a team of chemists to join his new company, Syntex. As Syntex grew, competitors emerged, and these efforts drove the cost of progesterone down from $80/gram to about $2/gram by the end of the 1940s.
Djerassi joined Syntex shortly after the war. With a ready supply of sex hormones established, work focused on developing synthetic analogues of progesterone that would mimic the biological activity but not be as easily metabolized. In 1951, Djerassi and coworkers Miramontes and Rozenkrantz developed the following route to a molecule called norethindrone. This molecule would eventually be the key ingredient of the birth control pills well into the 1970’s. [The whole story of Syntex, the Mexican steroid industry, and how the Pill was eventually developed and marketed would be a great subject of a movie – I’ve only excerpted a few parts. Djerassi’s book, Steroids Made It Possible is an interesting read]
OK. Let’s get to the chemistry.
6 Steps To The Pill
Djerassi’s route starts with the hormone estradiol, which is also readily obtained from the Mexican yam.
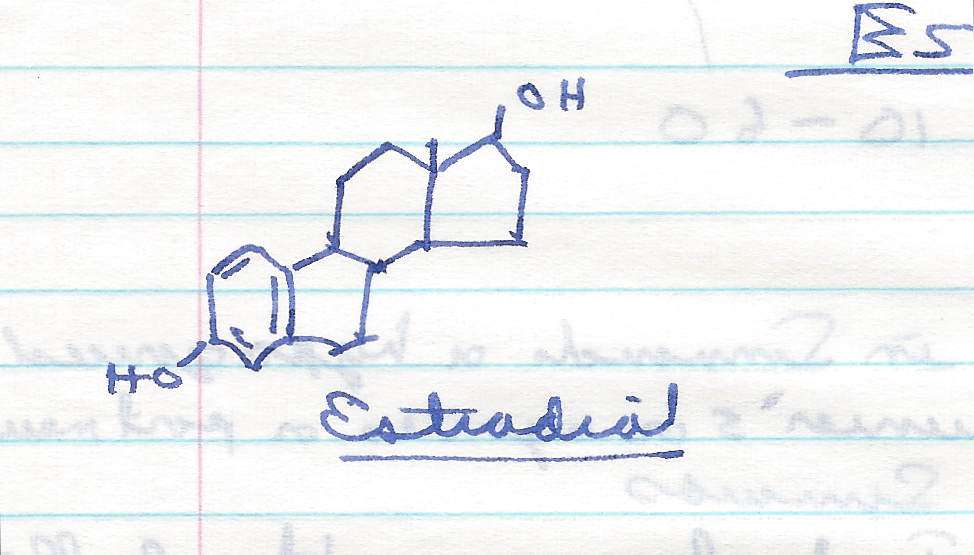
Norethindrone can be made in six steps from estradiol using the steps shown below.
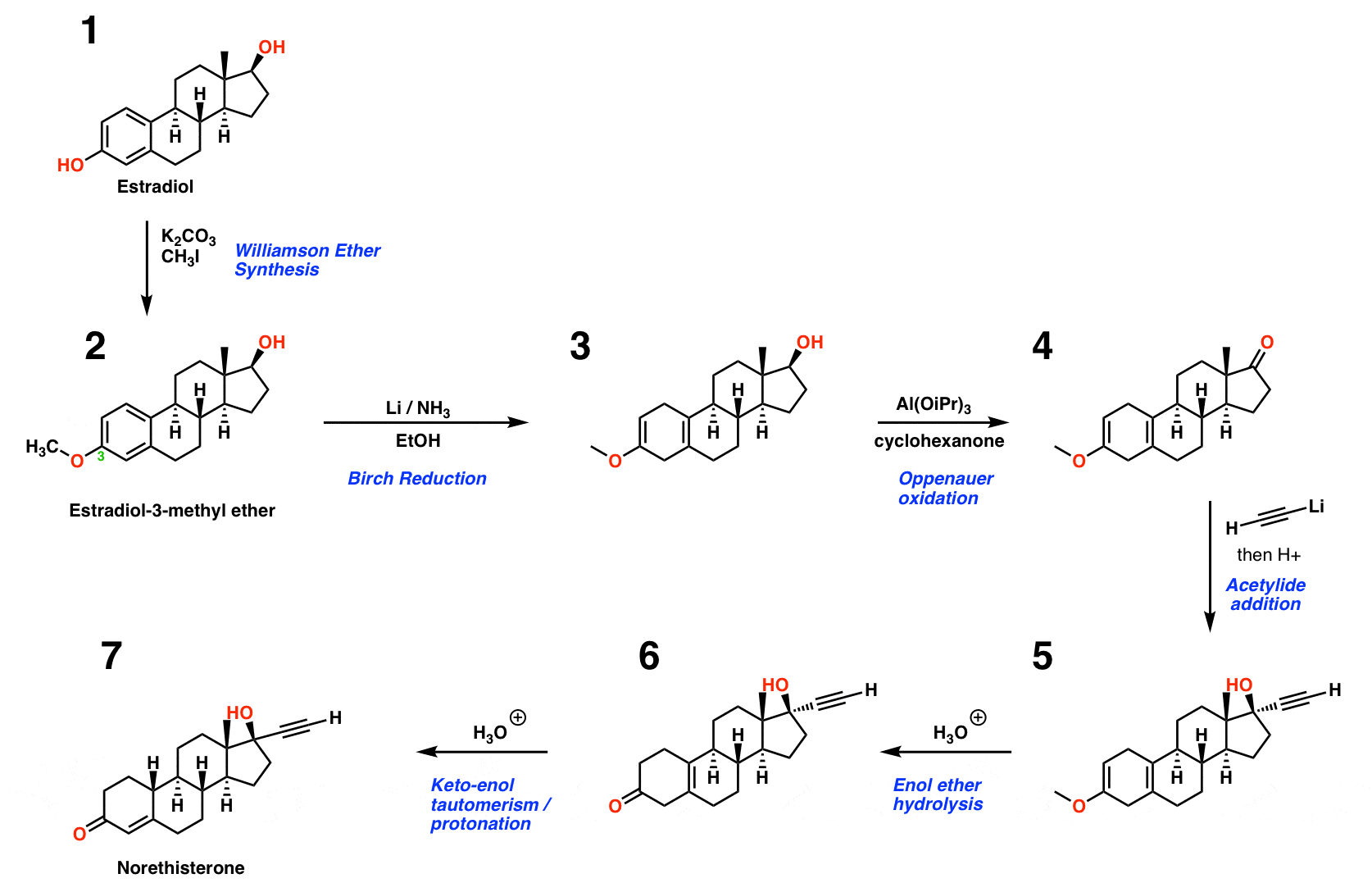
Step 1 – Williamson Ether
The first step is to make the 3-methyl ether of estradiol. [BTW: if you are wondering why that’s the “3” position, don’t worry about it. Seriously. Steroid numbering is weird.]
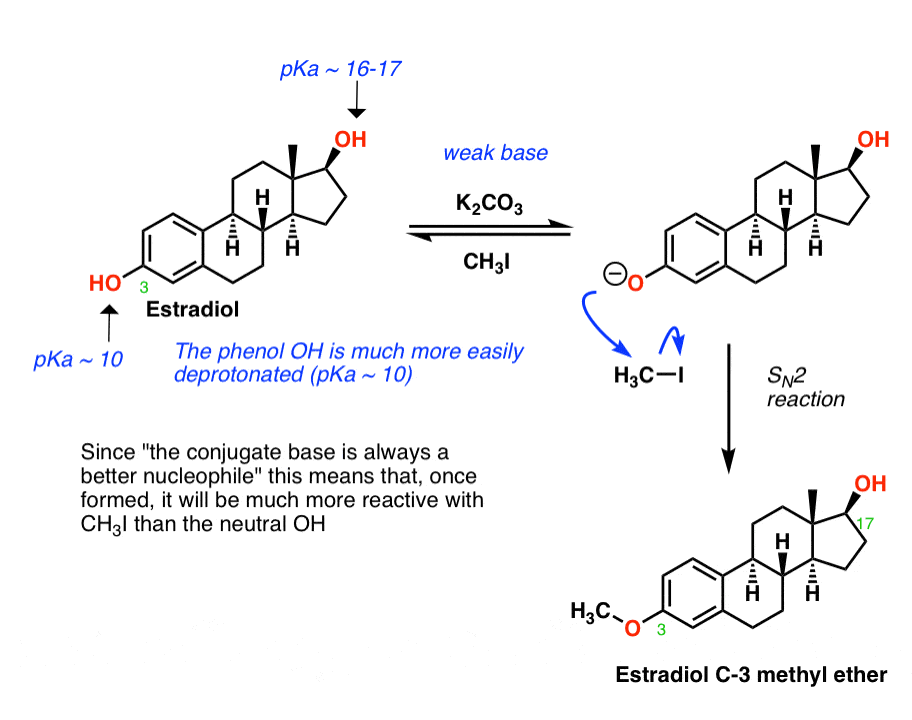
I’m not actually sure exactly which conditions Djerassi’s team used to do this, but I’ve shown one way it can be done via a Williamson Ether Synthesis. The idea is to use a weak base such as K2CO3 (potassium carbonate) in the presence of methyl iodide. Because the phenol OH is considerably more acidic than the secondary alcohol (pKa of 10 vs pKa of 17) it will be preferentially deprotonated by the weak base. Since the conjugate base is always a better nucleophile, the deprotonated phenol will react with the alkyl iodide, forming the ether.
Step 2 – Birch Reduction
The second step employs a Birch reduction to convert the aromatic ring into a diene. Because the OCH3 group is electron donating [through donation of the oxygen lone pairs], the reduction occurs in a way that maximizes the distance between the oxygen and the transient negative charge. The resulting product has a double bond with an OCH3 group attached, a functional group we call an enol ether. As we’ll see, this is a masked (or “protected”) form of a ketone.
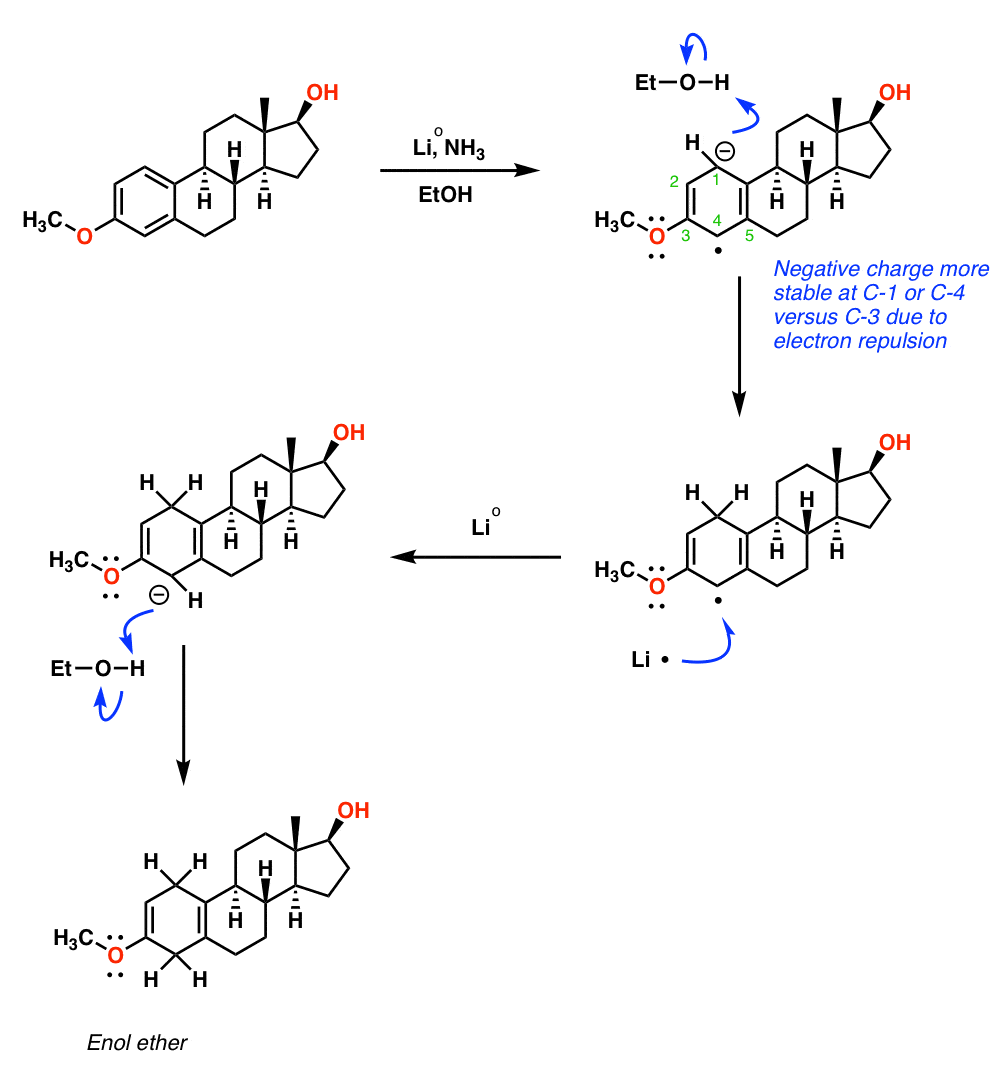
Step 3 – Oxidation of Secondary Alcohol
The next step is conversion of the secondary alcohol to a ketone. Djerassi’s route uses a reaction called the Oppenauer oxidation, which involves adding a Lewis acid catalyst such as Al(OiPr)3 in the presence of a ketone (usually acetone, but cyclohexanone was used here).
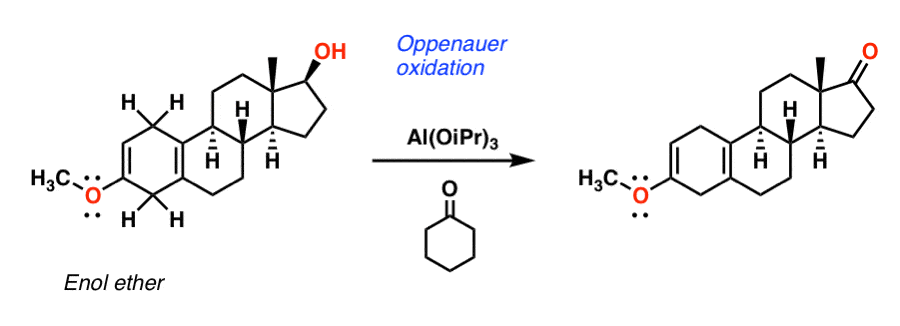
The key step involves a hydride transfer from the secondary alcohol to the ketone, resulting in the formation of cyclohexanol and the oxidized steroid. [If you learn the Canizarro reaction, the key step is similar].
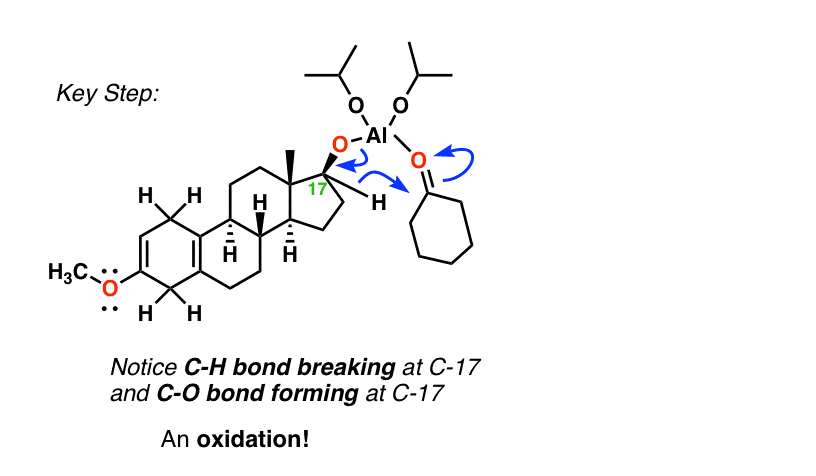
You might ask, “why not use PCC, or the Swern, or Dess-Martin to oxidize that alcohol? ” Great idea!! The thing is, this was done in 1951 and those reactions weren’t invented yet – but in retrospect, they’d work just as well. [One of the nice things about the Oppenauer is that it’s relatively cheap , an important factor when you’re making multiple kilograms of a product].
Step 4 – Addition of R-Li To A Ketone [essentially a Grignard reaction]
Ketones undergo a variety of addition reactions, notably from nucleophiles such as Grignard and organolithium reagents. In this step, lithium acetylide [the conjugate base of acetylene] is added to the ketone to form a tertiary alcohol (after quenching the resulting alkoxide with a weak acid). A subtlety of this particular reaction is that the acetylide approaches the ketone predominantly from the bottom face, resulting in the diastereomer shown. Any thoughts on why that might be?
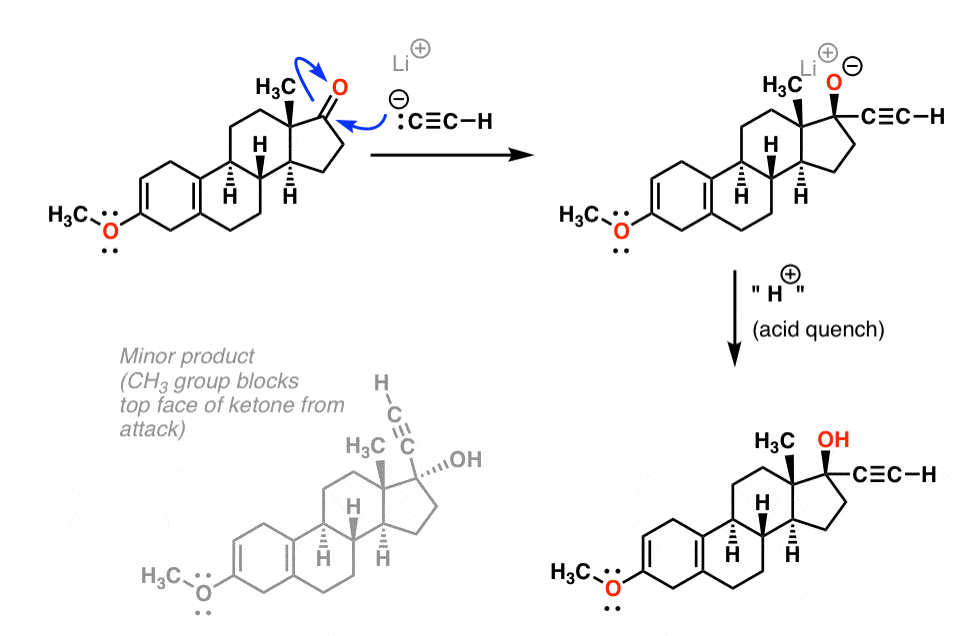
It’s the methyl group! That CH3 group serves to shield the ketone from attack from the top face, resulting in nearly exclusive approach of the nucleophile from the bottom.
Step 5 – Hydrolysis of the Enol Ether
In a previous step I alluded to the fact that the enol ether is a “masked” ketone. Adding aqueous acid converts the enol ether into a ketone.
We’ve discussed resonance and pi donation in previous posts. In the enol ether there is significant electron density on the end of the double bond (it is nucleophilic – try drawing a resonance form!). That means that it will react on the carbon with electrophiles such as acids. [See post: Reactions of Enols]
How does this reaction work? I’ve drawn out the mechanism here.
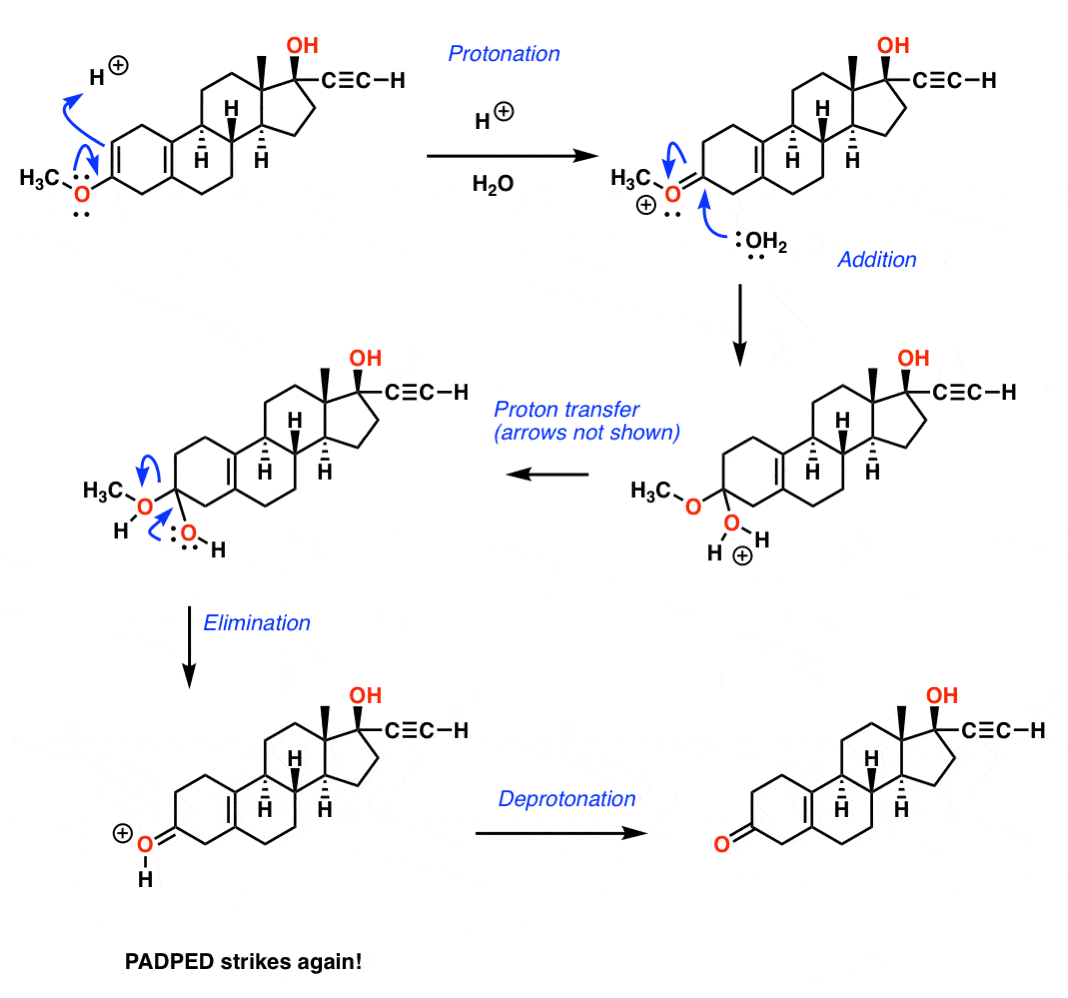
Protonation, addition of water, deprotonation of water, protonation of oxygen, elimination (of methanol), deprotonation. If that sounds vaguely familiar, you might recall that it’s yet another example of the PADPED pattern in action, a process I described in this video (making music with mechanisms).
Step 6 – Migration of the Double Bond
In the last step [in practice, this is just a continuation of the previous step] the double bond moves over one position so that it is in conjugation with the ketone. What’s going on here?
First of all, recall the ketone is in equilibrium with its tautomer, which is shown in the drawing below. Note that formation of the tautomer so that the double bond lies acoss C3-C4 results in an enol that is part of a conjugated diene.
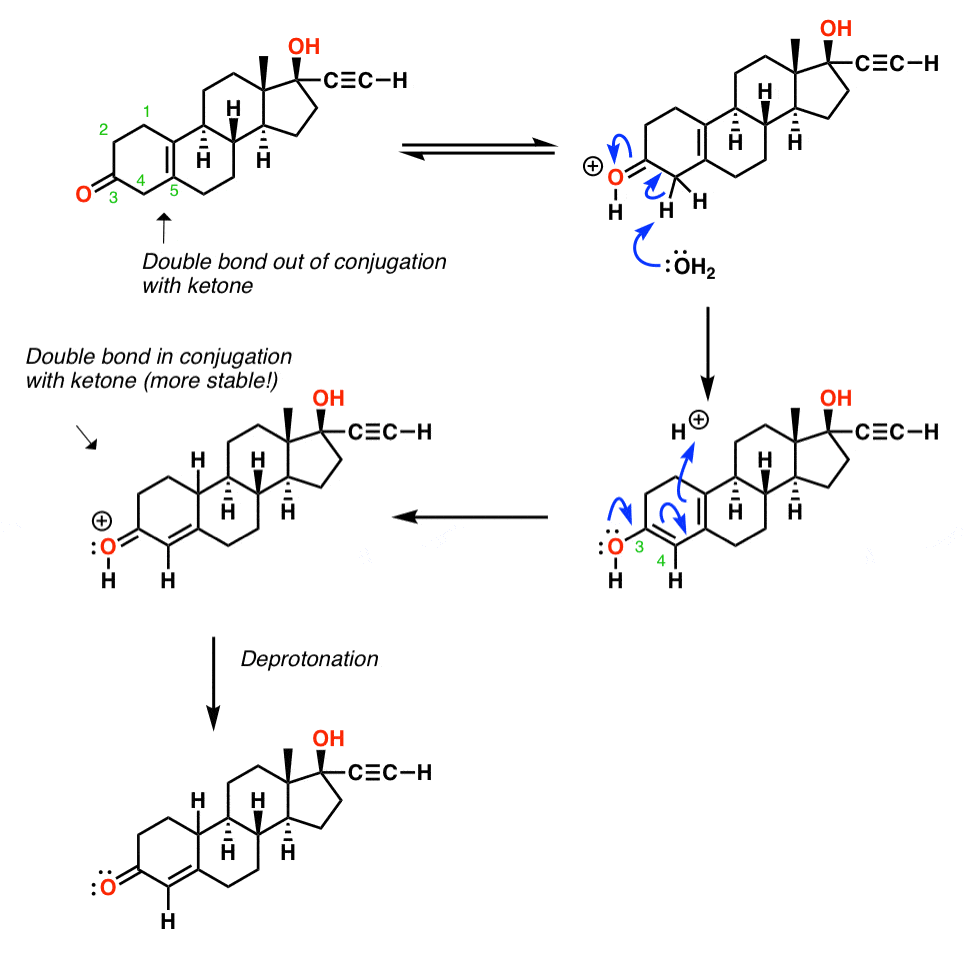
The protonation step is a classic example of “1,4” vs. “1,2” addition to a diene. When the proton is added to the end (the “4” position of the diene) and then revert back from the enol to the ketone, we end up with a situation where the ketone pi bond is in conjugation with the alkene pi bond. That situation is significantly more stable (about 2-3 kcal/mol) than that where the double bond lies out of conjugation.
The result is that the double bond has “migrated” into conjugation with the ketone.
Final Notes
One of the key elements in designing a good synthesis is ensuring that your reagent only reacts with the desired functional group, and this process is a perfect example of that.
Note how important the order of steps is. For example, it’s very important that the Birch reduction comes before the acetylide addition, because if it was done afterward, Li/NH3 might reduce the alkyne [recall Na/NH3 for instance]. It’s likewise important that the acetylide addition come before the unmasking of the enol ether, because otherwise the acetylide would end up adding to both ketones rather than just the one desired. That would lead to less of the desired product, and since Djerassi’s team was making a very precious compound, it would have had bad financial consequences as well. In that sense the enol ether acts as a protecting group for the leftmost ketone, which is only revealed after the key acetylide addition step.
So that’s the conversion of estradiol to norhindrone – state of the art for 1951, but all comprehensible to the dedicated student of organic chemistry who has gone about halfway through Org 2!
RIP Prof. Djerassi – may we all hope to have contributions that are so impactful to the lives of others.
R.I.P.
You know your made world history in chemistry when something you invent is called “The Pill.” R.I.P.