Bonding, Structure, and Resonance
How To Determine Hybridization: A Shortcut
Last updated: May 30th, 2026 |
A Shortcut For Determining The Hybridization Of An Atom In A Molecule
Here’s a shortcut for how to determine the hybridization of an atom in a molecule that will work in at least 95% of the cases you see in Org 1.
For a given atom:
- Count the number of atoms connected to it (atoms – not bonds!)
- Count the number of lone pairs attached to it.
- Add these two numbers together.
- If it’s 4, your atom is sp3.
- If it’s 3, your atom is sp2.
- If it’s 2, your atom is sp.
(If it’s 1, it’s probably hydrogen!)
The main exception is atoms with lone pairs that are adjacent to pi bonds, which we’ll discuss in detail below.
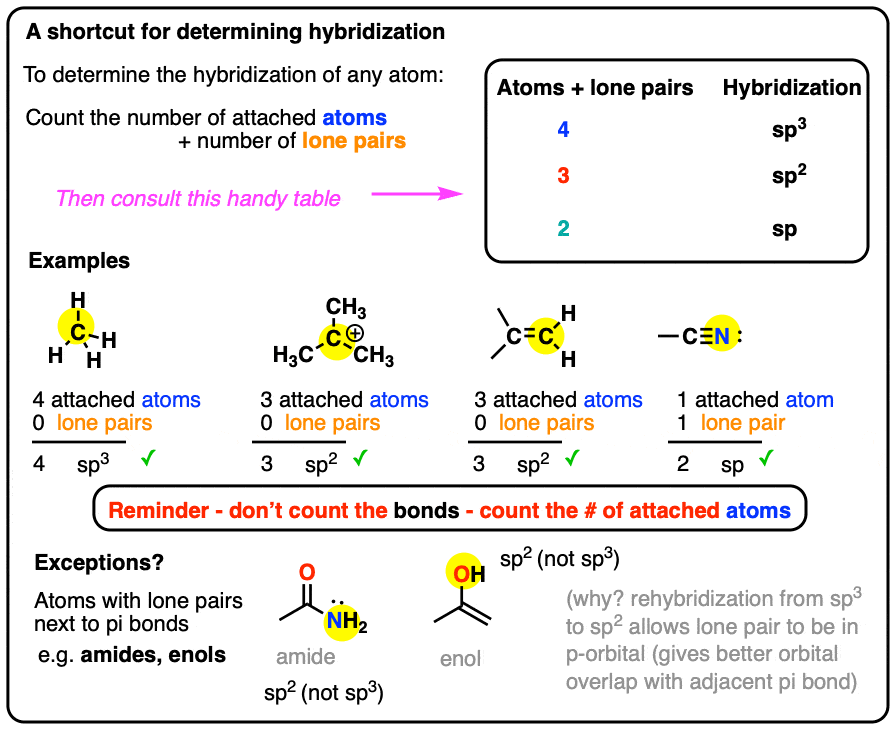
Table of Contents
- Some Simple Worked Examples Of The Hybridization Shortcut
- How To Determine Hybridization Of An Atom: Two Exercises
- Are There Any Exceptions?
- Exception #1: Lone Pairs Adjacent To Pi-bonds
- Lone Pairs In P-Orbitals (Versus Hybrid Orbitals) Have Better Orbital Overlap With Adjacent Pi Systems
- Exception #2. Geometric Constraints
- “Geometry Determines Hybridization, Not The Other Way Around”
- Notes
- Quiz Yourself!
1. Some Simple Worked Examples Of The Hybridization Shortcut
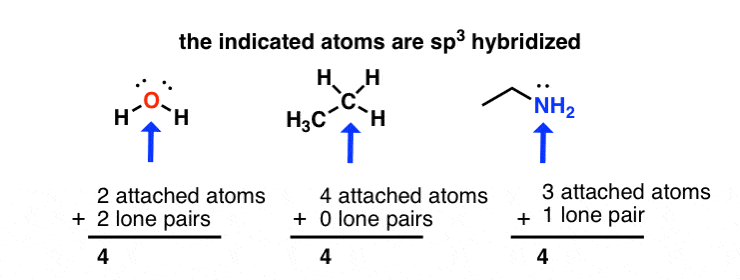
sp3 hybridization: sum of attached atoms + lone pairs = 4
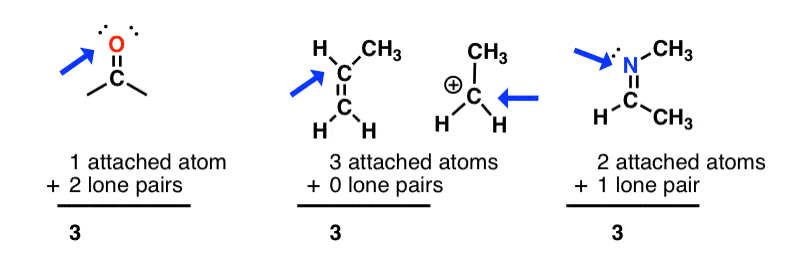
sp2 hybridization: sum of attached atoms + lone pairs = 3
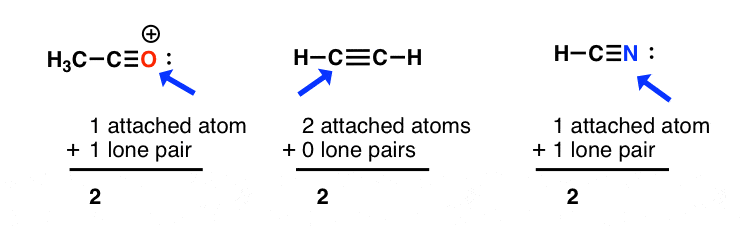
sp hybridization: sum of attached atoms + lone pairs = 2
Where it can start to get slightly tricky is in dealing with line diagrams containing implicit (“hidden”) hydrogens and lone pairs.
Chemists like time-saving shortcuts just as much as anybody else, and learning to quickly interpret line diagrams is as fundamental to organic chemistry as learning the alphabet is to written English.
Remember:
- Just because lone pairs aren’t drawn in on oxygen, nitrogen, and fluorine doesn’t mean they’re not there.
- Assume a full octet for C, N, O, and F with the following one exception: a positive charge on carbon indicates that there are only six electrons around it. [Nitrogen and oxygen bearing a formal charge of +1 still have full octets].
[Advanced: Note 1 covers how to determine the hybridization of atoms in some weird cases like free radicals, carbenes and nitrenes ]
2. How To Determine Hybridization Of An Atom: Two Exercises
Here’s an exercise. Try picking out the hybridization of the atoms in this highly poisonous molecule made by the frog in funky looking pyjamas, below right.
[Don’t worry if the molecule looks a little crazy: just focus on the individual atoms that the arrows point to (A, B, C, D, E). A and B especially. If you haven’t mastered line diagrams yet (and “hidden” hydrogens) maybe get some more practice and come back to this later.]
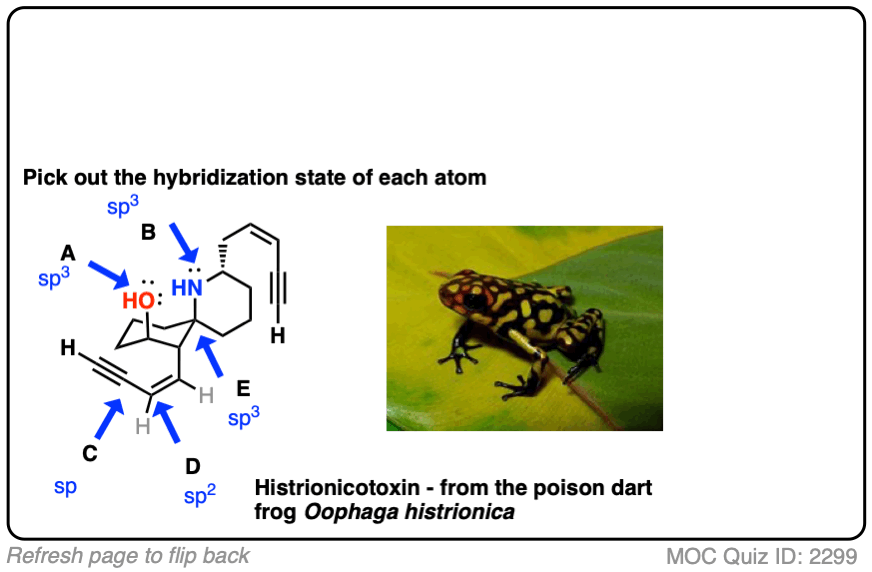
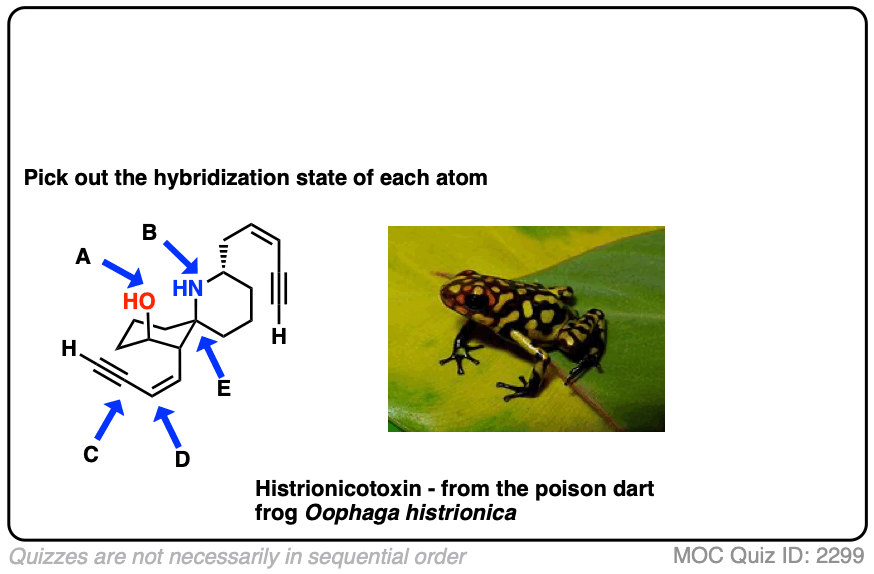 Click to Flip
Click to Flip
Here are some more examples.
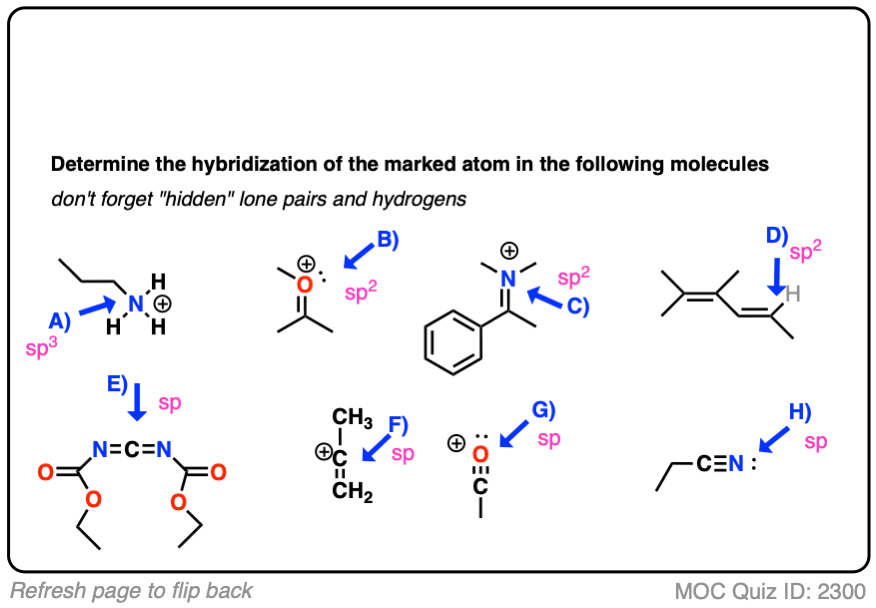
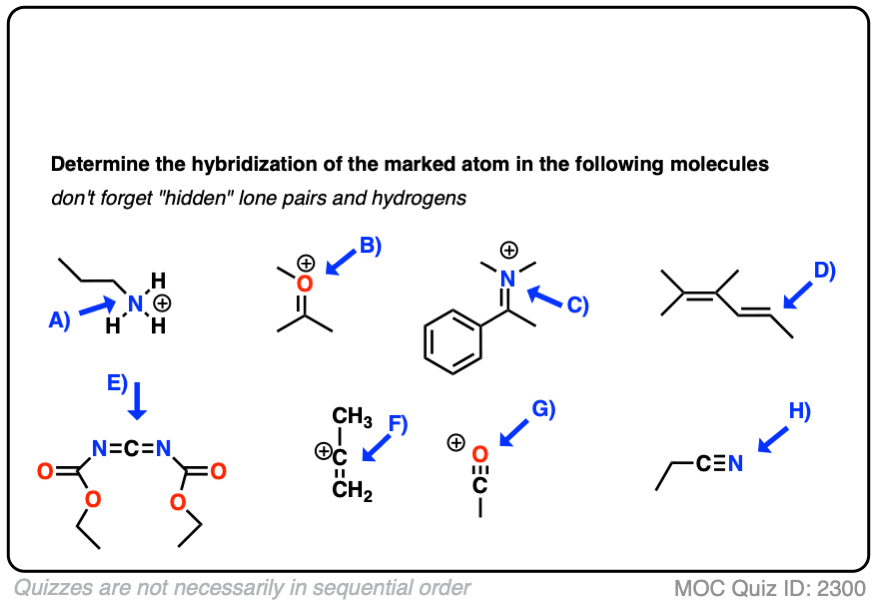 Click to Flip
Click to Flip
More practice quizzes for hybridization can be found here (MOC Membership unlocks them all)
3. Are There Any Exceptions?
Sure. Although as with many things, explaining the shortcut takes about 2 minutes, while explaining the exceptions takes about 10 times longer.
Helpfully, these exceptions fall into two main categories. It should be noted that by the time your course explains why these examples are exceptions, it will likely have moved far beyond hybridization. Phytoplankton
Bottom line: these probably won’t be found on your first midterm.
4. Exception #1: Lone Pairs Adjacent To Pi-bonds
The main exception is for atoms bearing lone pairs that are adjacent to pi bonds.
Quick shortcut: Lone pairs adjacent to pi-bonds (and pi-systems) tend to be in unhybridized p orbitals, rather than in hybridized spn orbitals.
This is most common for nitrogen and oxygen.
In the cases below, a nitrogen or oxygen that we might expect to be sp3 hybridized is actually sp2 hybridized (trigonal planar).
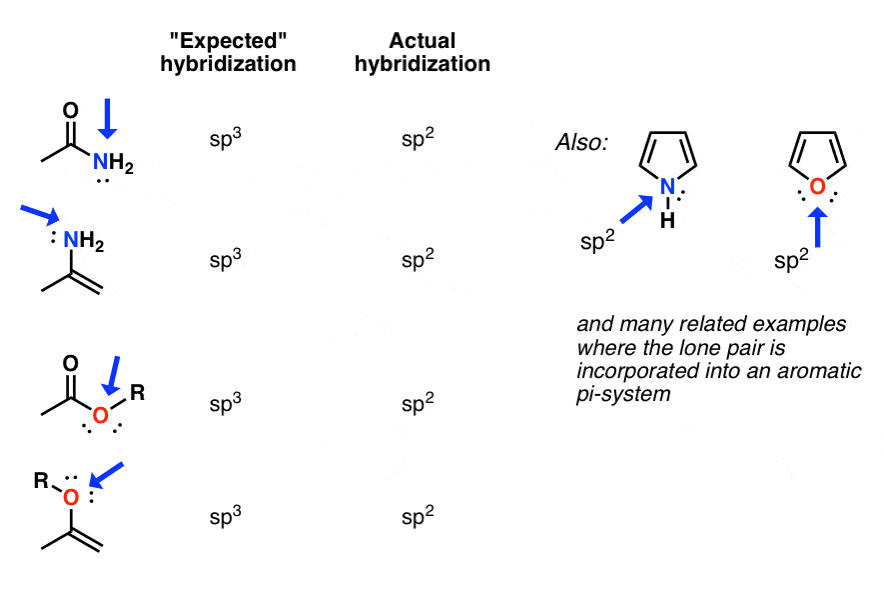
Why? The quick answer is that lowering of energy from conjugation of the p-orbital with the adjacent pi-bond more than compensates for the rise in energy due to greater electron-pair repulsion for sp2 versus sp3
[see this post: “Conjugation and Resonance“]
What’s the long answer?
5. Lone Pairs In P-Orbitals (Versus Hybrid Orbitals) Have Better Orbital Overlap With Adjacent Pi Systems
Let’s think back to why atoms hybridize in the first place: minimization of electron-pair repulsion.
For a primary amine like methylamine, adoption of a tetrahedral (sp3) geometry by nitrogen versus a trigonal planar (sp2) geometry is worth about 5 kcal/mol [roughly 20 kJ/mol].
That might not sound like a lot, but for two species in equilibrium, a difference of 5 kcal/mol in energy represents a ratio of about 4400:1 . [How do we know this? See this (advanced) Note 2 on nitrogen inversion]
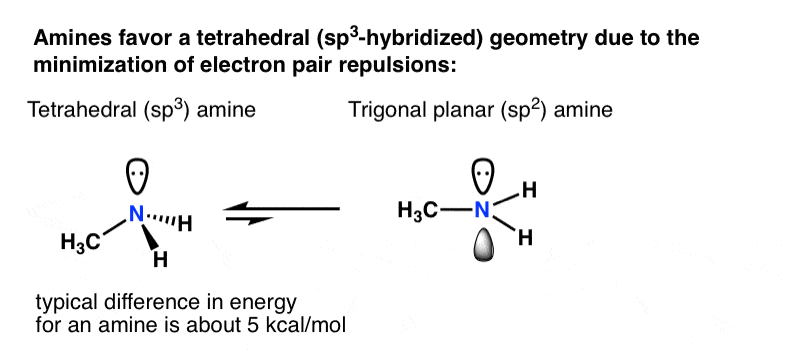
What if there was some compensating effect whereby a lone pair in an unhybridized p-orbital was actually more stable than if it was in a hybridized orbital?
This turns out to be the case in many situations where the lone pair is adjacent to a pi bond! The most common and important example is that of amides, which constitute the linkages between amino acids. The nitrogen in amides is planar (sp2), not trigonal pyramidal (sp3), as proven by X-ray crystallography.
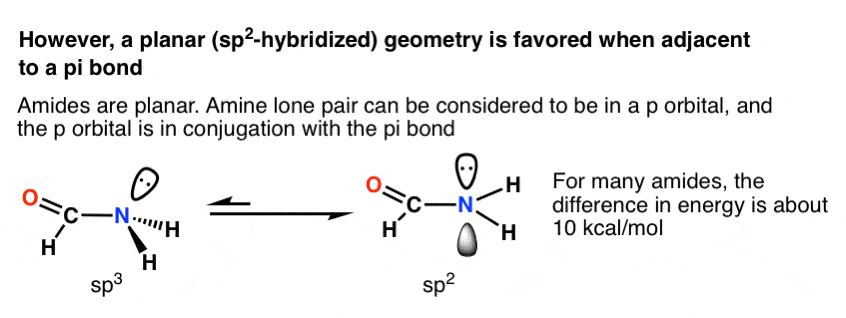
The difference in energy varies widely, but a typical value is about 10 kcal/mol favouring the trigonal planar geometry. [We know this because many amides have a measurable barrier to rotation – a topic we also talked about in the Conjugation and Resonance post]
Why is trigonal planar geometry favoured here? Better orbital overlap of the p orbital with the pi bond vs. the (hybridized) sp3 orbital.
The drawing below tries to show how a change in hybridization from sp3 to sp2 brings the p-orbital closer to the adjoining p-orbitals of the pi bond, allowing for better orbital overlap. Better orbital overlap allows for stronger pi-bonding between the nitrogen lone pair and the carbonyl p-orbital, which results in an overall lowering of energy. 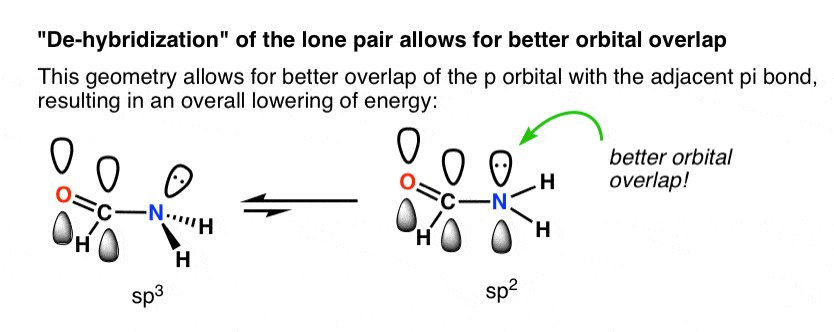
You can think of this as leading to a stronger “partial” C–N bond. Two important consequences of this interaction are restricted rotation in amides, as well as the fact that acid reacts with amides on the oxygen, not the nitrogen lone pair (!)
The oxygen in esters and enols is also also sp2 hybridized, as is the nitrogen in enamines and countless other examples.
As you will likely see in Org 2, some of the most dramatic cases are those where the “de-hybridized” lone pair participates in an aromatic system. Here, the energetic compensation for a change in hybridization from sp3 to sp2 can be very great indeed – more than 20 kcal/mol in some cases.
For this reason, the most basic site of pyrrole is not the nitrogen lone pair, but on the carbon (C-2) (!).
6. Exception #2. Geometric Constraints
Another example where the actual hybridization differs from what we might expect from the shortcut is in cases with geometric constraints. For instance in the phenyl cation below, the indicated carbon is attached to two atoms and zero lone pairs.
What’s the hybridization?
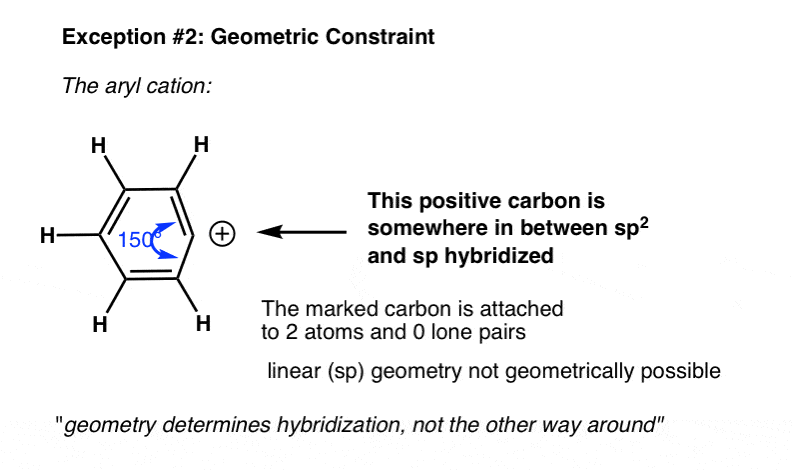
{kind=link}
From our shortcut, we might expect the hybridization to be sp.
In fact, the geometry around the atom is much closer to sp2. That’s because the angle strain adopting the linear (sp) geometry would lead to far too much angle strain to be a stable molecule.
7. “Geometry Determines Hybridization, Not The Other Way Around”
A quote passed on to me from Matt seems appropriate:
“Geometry determines hybridization, not the other way around”
Well, that’s probably more than you wanted to know about how to determine the hybridization of atoms.
Suffice to say, any post from this site that contains shortcut in the title is a sure fire-bet to have over 1000 words and >10 figures.
Thanks to Matt Pierce of Organic Chemistry Solutions for important contributions to this post. Ask Matt about scheduling an online tutoring session.
Notes
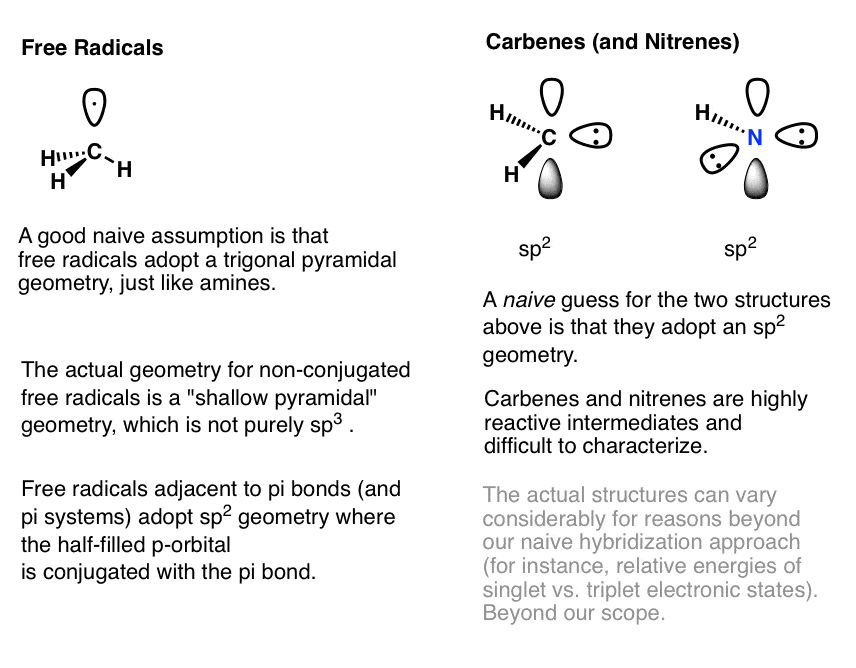
Sometimes you might be asked to determine the hybridization of free radicals and of carbenes (or nitrenes)
Although you’re unlikely to encounter these, let’s still have a look.
- Free radicals exist in a shallow pyramidal geometry, not purely sp2 or sp3.
- However, if they are adjacent to a pi system (e.g. a C-C double or triple bond) then the shallow pyramid will re-hybridize to give it an sp2 geometry, which allows for full resonance delocalization of the free radical.
- Carbenes and nitrenes would give us sp2 geometry by the hybridization shortcut. However their actual structures can vary depending on whether or not the electron pair exists in a single orbital (a singlet carbene) or is divided into two singly-filled orbitals (a triplet carbene). That’s really beyond the scope of introductory organic chemistry.
What about higher block elements like sulfur and phosphorus?
Third row elements like phosphorus and sulfur can exceed an octet of electrons by incorporating d-orbitals in the hybrid. This is more in the realm of inorganic chemistry so I don’t really want to discuss it. Here’s an example for the hybridization of SF4 from elsewhere. (sp3d orbitals).
Note 2: For the 5 kcal/mol figure, see here. [Tetrahedron Lett, 1971, 37, 3437]. (Kurt Mislow, RIP. )
An amine connected to three different substituents (R1 R2 and R3) should be chiral, since it has in total 4 different substituents (including the lone pair). However, all early attempts to prepare enantiomerically pure amines met with failure. It was later found that amines undergo inversion at room temperature, like an umbrella being forced inside-out by a strong wind.
In the transition state for inversion the nitrogen is trigonal planar. One can thus calculate the difference in energy between the sp3 and sp2 geometries by measuring the activation barrier for this process (see ref).
Note 3:A fun counter-example might be Coelenterazine .
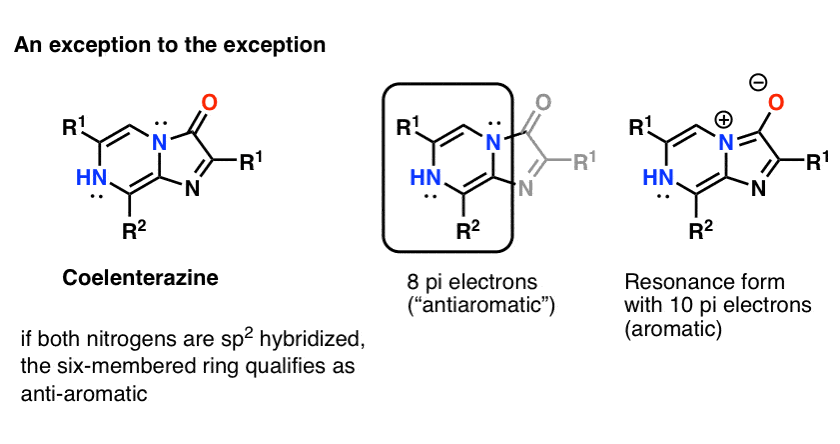
One would not expect both nitrogen atoms to be sp2 hybridized, because that would lead to a cyclic, flat, conjugated system with 8 pi electrons : in other words, antiaromatic. I can’t find a crystal structure of the core molecule to confirm (but would welcome any additional information!)
NOTE – (added afterwards) If you draw the resonance form where the nitrogen lone pair forms a pi bond with the carbonyl carbon, then the ring system has 10 electrons and would therefore be “aromatic”.
Quiz Yourself!
Become a MOC member to see the clickable quiz with answers on the back.
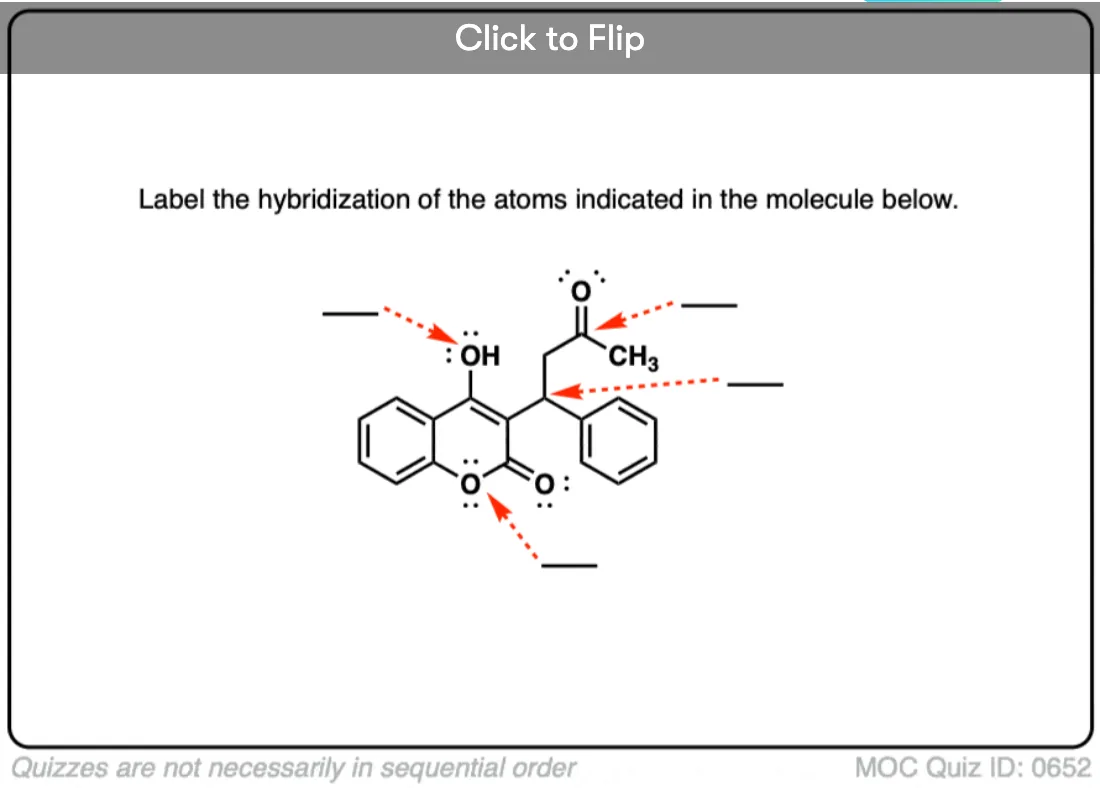
Become a MOC member to see the clickable quiz with answers on the back.

Become a MOC member to see the clickable quiz with answers on the back.

Become a MOC member to see the clickable quiz with answers on the back.
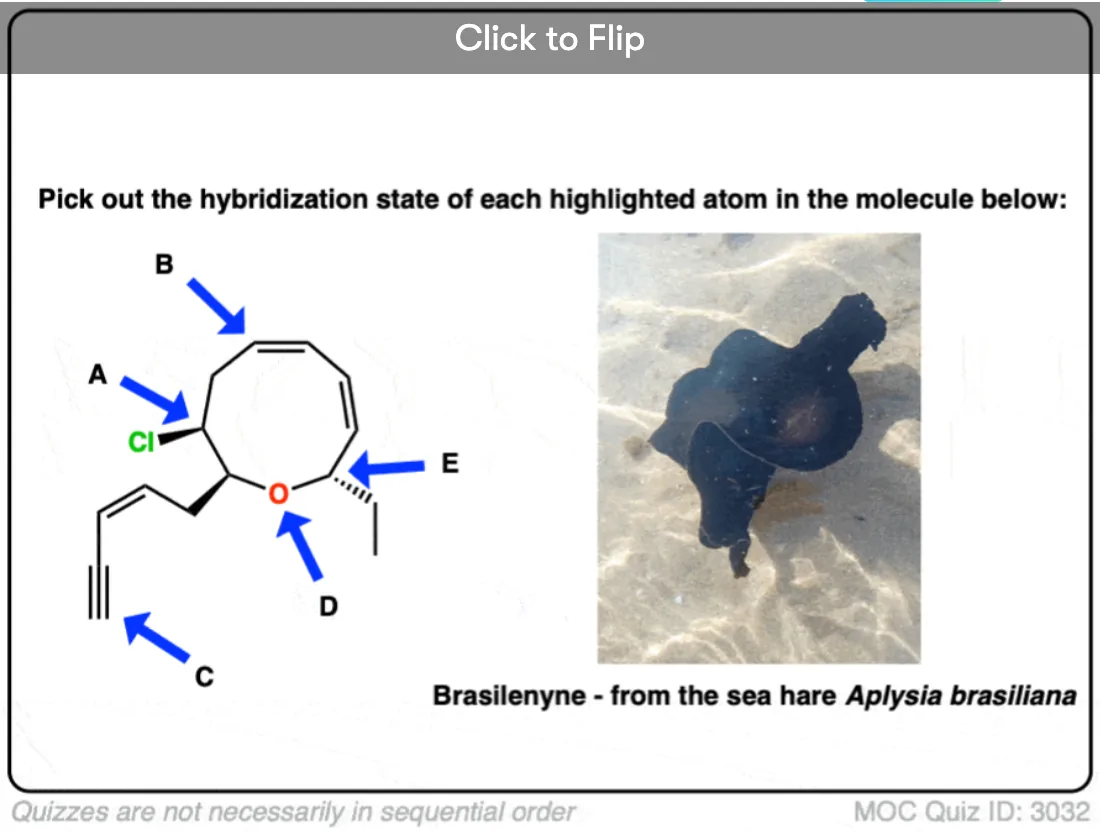
Become a MOC member to see the clickable quiz with answers on the back.
- Barrier to pyramidal inversion of nitrogen in dibenzylmethylamine
Michael J. S. Dewar and W. Brian Jennings
Journal of the American Chemical Society 1971 93 (2), 401-403
DOI: 10.1021/ja00731a016 -
Pyramidal inversion barriers: the significance of ground state geometry
Joseph Stackhouse, Raymond D.Baechler, Kurt Mislow
Tetrahedron Letters Volume 12, Issue 37, 1971, Pages 3437-3440
DOI: doi.org/10.1016/S0040-4039(01)97199-0
Thanks for the explanation.But am confused 🤔🤔”what’s the difference between attached atoms and lone pairs???”
Henry Ugbomah;
The view that geometry determines hybridization, and not otherwise is quite taken. Amine enantiomers are unsuccessful because, at room temperature, they undergo inversion is also interesting. However, the literature states, that catalysts can induce hybridization, by changing the electronic environment surrounding atoms.
The direction of the equal sign may be wrong in the image below this sentence.
Better orbital overlap allows for stronger pi-bonding between the nitrogen lone pair and the carbonyl p-orbital, which results in an overall lowering of energy.
i am in love with this site….. explains so clear and good….. really man i was irritated because i was not able to understand but now i understand it just because of this site….
Sir thankyou so much for your explain , i was able to get a lot of things I couldn’t understand earlier, thanks a lot ,but I have a doubt about the last note ….how did it go from antiaromatic to aromatic in coelentrazine
Thank you for the great post, as usual.
I think there is a typo in the first section of point 5 and its picture; the geometry is trigonal pyramidal instead of tetrahedral.
very helpful thanks a lot SIR
thank youuuuu!!! your discussions really helped my laboratory report in organic chemistry which is due tomorrow. can you have a discussion about the effect of pi systems? i’m looking forward to it!
may i ask for the carbon hybridization of pentane and the effects of the pi system.
Pentane, C5H12 ? Tetrahedral carbons, sp3 hybridized
Well I don’t know what to say is was really helpful to I was able to understand it thank u very much
Hi James! Firstly, thank you so much for your explanation of hybridization! Here I have a question. It says if atom with lone pair next to pi bond, rehydridization will occur so we cannot use the instruction above. So how can we determine whether an atom with lone pair is next to pi bond. Thank you!
Hi, most familiar example would be the lone pairs on the OH group of a carboxylic acid, R-CO2H. The OH oxygen is sp2 hybridized since the lone pair is on an atom adjacent to a pi bond (i.e. the C=O pi bond).
Another example would be an ester, R-CO2CH3 . In this case the lone pair on the oxygen bearing the CH3 (i.e. O-CH3) is sp2 hybridized since it is adjacent to a C=O bond.
Amides, R-C(O)-NH2 have sp2-hybridized nitrogens, since the lone pair on the nitrogen is adjacent to a C=O bond.
The negatively charged carbon in the “allyl anion” is sp2 hybridized.
Lots more examples but these are a few.
This was a great review! I hadn’t done nitrogen hybridization in years and needed a quick refresher. Thanks!!!
Thanks Karla
How to determine the hybridisation state of N atom number 3 in this imidazole ring diagram?: https://upload.wikimedia.org/wikipedia/commons/thumb/b/b8/Imidazole_2D_numbered.svg/110px-Imidazole_2D_numbered.svg.png
Since by geometry, you would expect it to be sp2 hybridised, but there’s also an adjacent pi-bond system (C4 and C5), so you would expect it to be sp hybridised (such that the lone pair occupies p orbital and can undergo resonance with C=C pi orbital).
The art of teaching is so wonderful. Very clear and step-by-step explanations. My heartfelt thanks to you. My congratulations on continuing your service further.
I wonder what will be hybridization on carbocation of ethynylium ion or ethynyl carbocation.
Hi James, thanks for the concise and straight forward explanation of these exceptions. I’m teaching an orgo course this fall and feel better prepared to explain this to students.
Glad to hear it, CK, glad you find it helpful.
Hi! First, I’d like to say that I find your posts extremely helpful, certainly most of the tricks in organic chemistry I’ve learned in here. Reading this post and studying the subject I was thinking about the azobenzene and hydrazobenzene structures, I’d expect them to be sp2 and sp3, respectively, but since they have benzene rings connected to each nitrogen, would these hybridizations be valid?
Hello, thank you so much for the in-depth explanation. I’d just like to ask if exception #1 (Lone Pairs Adjacent To Pi-bonds) applies to N atom of HCN? It’s an example included in the first section as sp, but I would just like to clarify since the resources I’ve found are conflicting. Hehe, again thanks so much, sir! I hope you are well and safe.
The N atom of HCN is sp hybridized. One sp orbital is the C-N sigma bond, and the other has the lone pair on nitrogen. Under no conditions does the lone pair on nitrogen participate in resonance, since that would result in a nitrogen species with six electrons around it (less than an octet) which is very unstable!
What if the number of atom connected to it and the lone pair whe added is more than four, in total what do u call such type of hybridization
I would be wary of applying hybridization concepts to bonds in the 3rd row, such as sulfur, that exceed a full octet.
Time saving concept
Thanks a lot for this info. I searched everywhere but could not get anything on these exceptions of hybridization.
Glad you found it helpful!
very helpful! thanks:))
Glad you found it helpful!
Thanks for a clear explanation of why N and O atoms next to a pi bond or system would rather be sp2 hybridized. Gives me deeper insight as a non – organic chem teacher.
You are welcome Willetta. Thanks for stopping by.
Thanks again! I am finally gaining some facility at this thanks to your deep understanding coupled with your very clear writing.
Glad to hear it John. Thank you.
You, sir, are a tremendous help and credit to the profession of education. Thank you, thank you!
The 1H-NMR of coelenterazine (DOI: 10.1021/ct300356j) shows two signals at 9.13 (s, 1 H), and 6.44 (bs, 1 H), which suggests that the system is conjugated with the carbonyl making it all planar and aromatic when considering the entire bicyclic system. You can observe similar deshielding effects in, say, azulene for the protons on the 7-membered ring.
Now that I look at it again, you’re absolutely right. Thanks Victor.
“Third row elements like phosphorus and sulfur can exceed an octet of electrons by incorporating d-orbitals in the hybrid.”
This is incorrect, and was proven wrong years ago. See http://pubs.acs.org/doi/abs/10.1021/ja00273a006 and citing references therein. It’s more accurate (and more intuitive) to continue to follow the octet rule for sulfur, phosphorus, and other heavy main group elements. SF4, for example, can be represented as four equal-weight resonance structures of the form [SF3]+[F]-, giving an overall bond order of 0.75 for each S-F bond. This way, every atom follows the octet rule in each resonance structure. Of course you could always use molecular orbital theory in conjunction with symmetry-adapted linear combination of atomic orbitals, and then you wouldn’t need to deal with “expanded octets” in hypercoordinate molecules.
Yes and no.
There’s nothing intrinsically wrong in the phrase itself as the “hypervalent” atoms DO use the higher orbitals to some extent. It is more to the point of what orbitals are involved in the overall bonding scheme. And no, nobody, who has at least some understanding of the concept of the hybridization, will insist that by saying that sulfur in SF4 has the sp3d hybridization will strictly mean that we have 100% involvement of 1 s, 3 p, and 1 d orbital in the bonding structure.
It’s the same kind of argument we can bring when discussing, say, cyclopropanone. What is the hybridization of the carbonyl carbon there? Is it sp2? Is it sp2+? Is it sp2-? Is it somewhere in between? What about the hybridization in di-central rhenium complexes with quaternary bond? Or riddle me out, for instance, the exact iodine’s hybridization in every form of periodic acid ;)
When we acknowledge the limitations of the theories we use, they are in a pretty good agreement with each other ;) And while using the MO is the best way to go, it is not what is being taught at the general chemistry or organic chemistry level, nor it is what students are facing on the test.