Spectroscopy
Natural Product Isolation (2): Purification Techniques, An Overview
Last updated: June 2nd, 2026 |
How to obtain a pure compound from a crude mixture: 5 Key Purification Techniques
Previously, we showed a few examples of preparing crude extracts from plants and other organisms. While sometimes (rarely) we get lucky and obtain an extract that is predominantly one compound, a more representative situation is that a mixture of compounds is obtained. For instance, this gas chromatography (GC) analysis of lavender oil says it all, with 36 marked compounds (and more than that if you scour the baseline).
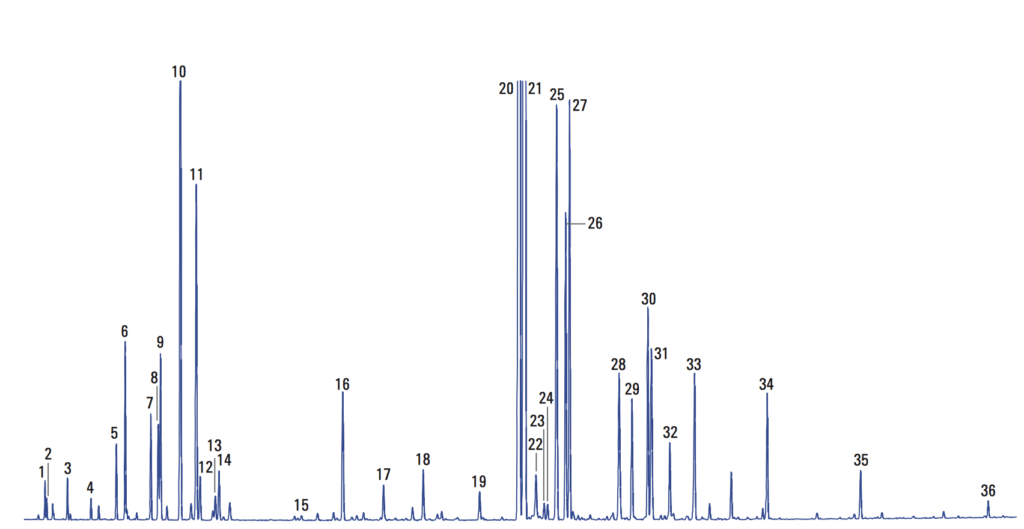
In this post, we’ll go through some fundamental techniques for separation and purification of crude mixtures, as one might obtain from natural product extracts.
The main question we want to answer is this: what are some options for purifying a crude mixture into its components?
There are five key techniques we’ll cover today. Let’s go!
Table of Contents
- Taking Advantage of Chemical Properties (Acid-Base)
- Separation By Boiling Point Differences (Distillation)
- Crystallization
- Chromatography
- Gas Chromatography (GC) and High-Performance Liquid Chromatography (HPLC)
- Conclusion: Purification Techniques
- Notes
1. Chemical Properties (Acid-Base)
One of the oldest (and still widely used) methods for natural product isolation is to modify the pH, and thus the water-solubility, of acidic and basic molecules in the mixture. This is because certain basic and acidic molecules can easily be converted into salts, which greatly increases their water solubility. This process is generally reversible, so that after the charged salt is separated from the rest of the mixture, we can recover the original acidic or basic species.
Here’s a general overview.
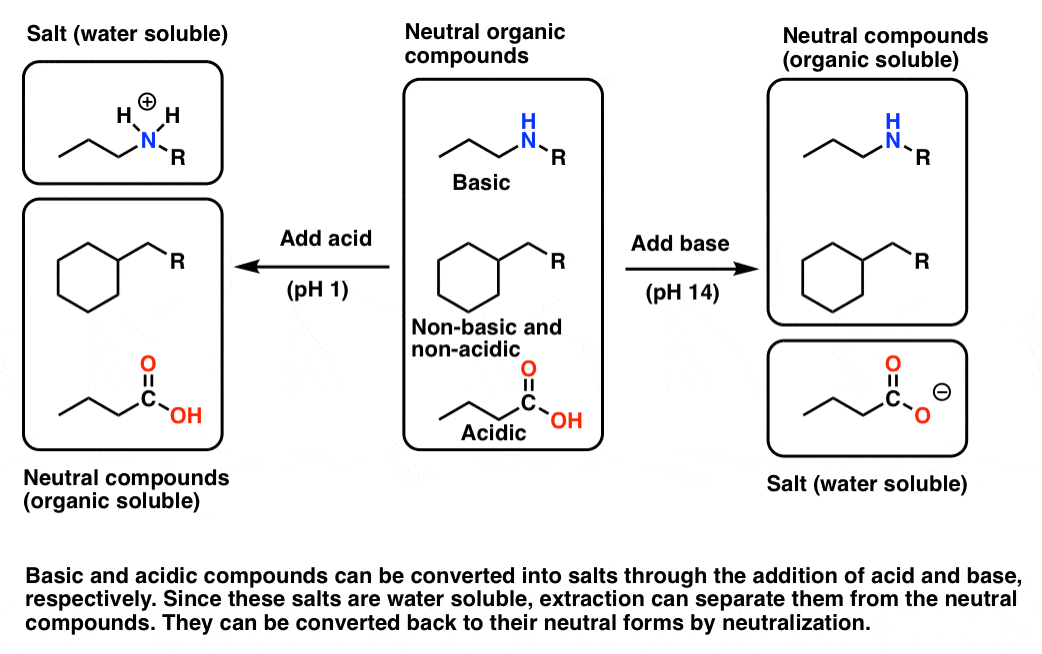
Let’s say your crude mixture has a molecule that can act as a base – an amine, for example. In their neutral form, most amines are only soluble in organic solvents such as diethyl ether or dichloromethane. However, if one adjusts the pH down to about 1 or so, the amine will be protonated, forming an ammonium salt (its conjugate acid). The salt, now bearing a charge, will then have considerable water solubility, so extraction of the mixture with water will separate the salt from the crude mixture. The aqueous phase can then be collected, and the pH adjusted back to neutral through the addition of base. This will neutralize the acid, causing precipitation of the amine out of the aqueous phase. The neutral amine can be extracted with an organic solvent.
Here’s a representative scheme (click to embiggen)
The class of natural products called alkaloids were among the earliest to be isolated as pure molecules due to their acid base properties. For example, here is a painting of the German chemist Friederich Sertürner hanging out with his buddies after isolating morphine in 1824. [This is artist Robert Thom’s reimagining of the scene, by the way, not a posed portrait.]

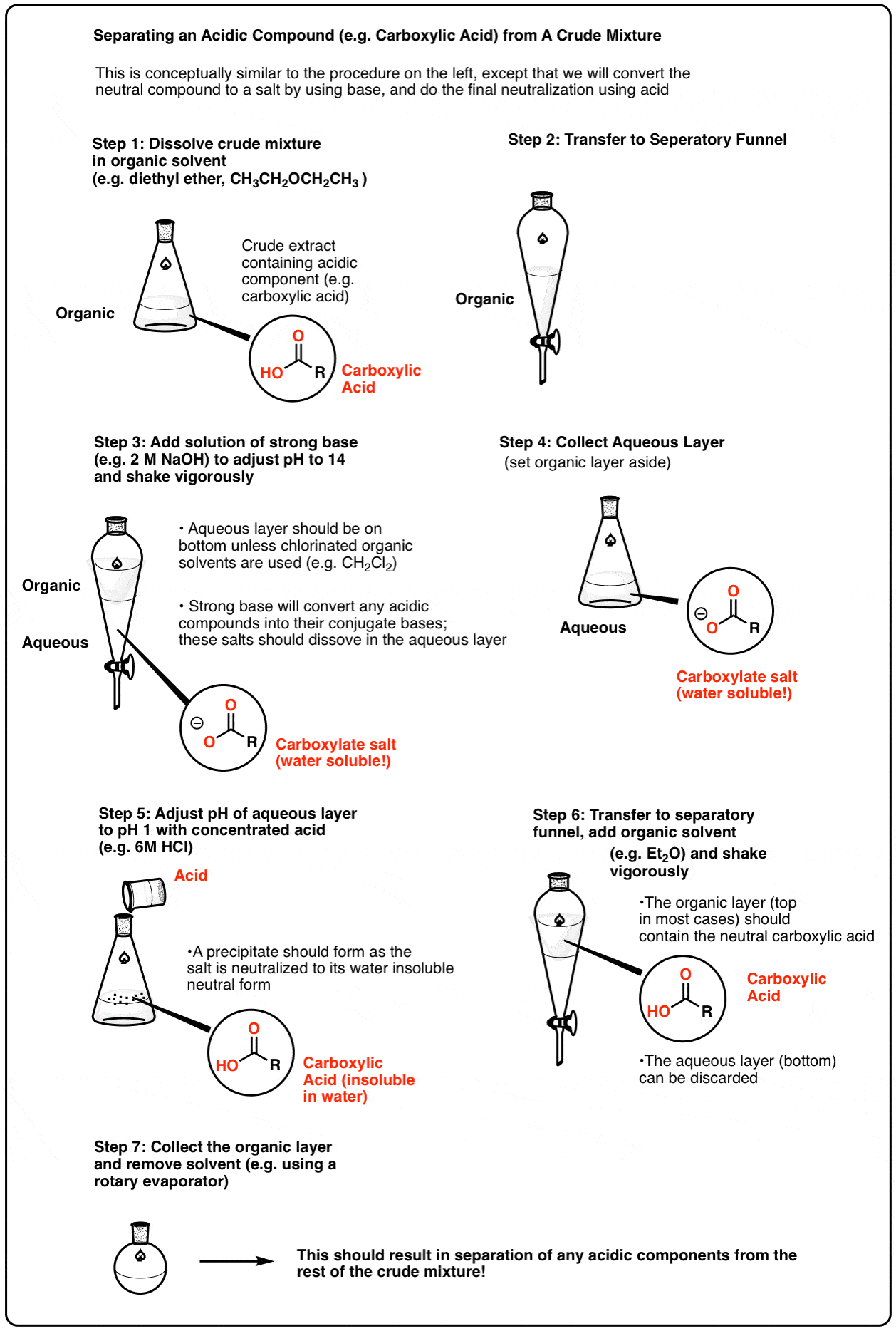
This also works the opposite way, for acidic components. For example, one can dissolve the crude extract in an organic solvent and then extract with strong base (pH 14). Any components with acidic functional groups (such as carboxylic acids) will be converted into their conjugate bases, and the resulting salts will be relatively water-soluble. The aqueous extract can then be removed, and then re-acidified in order to regenerate the parent compound. Extraction with an organic solvent then results in separation of the acid.
Here’s a representative scheme (click to enlarge). PDF Version
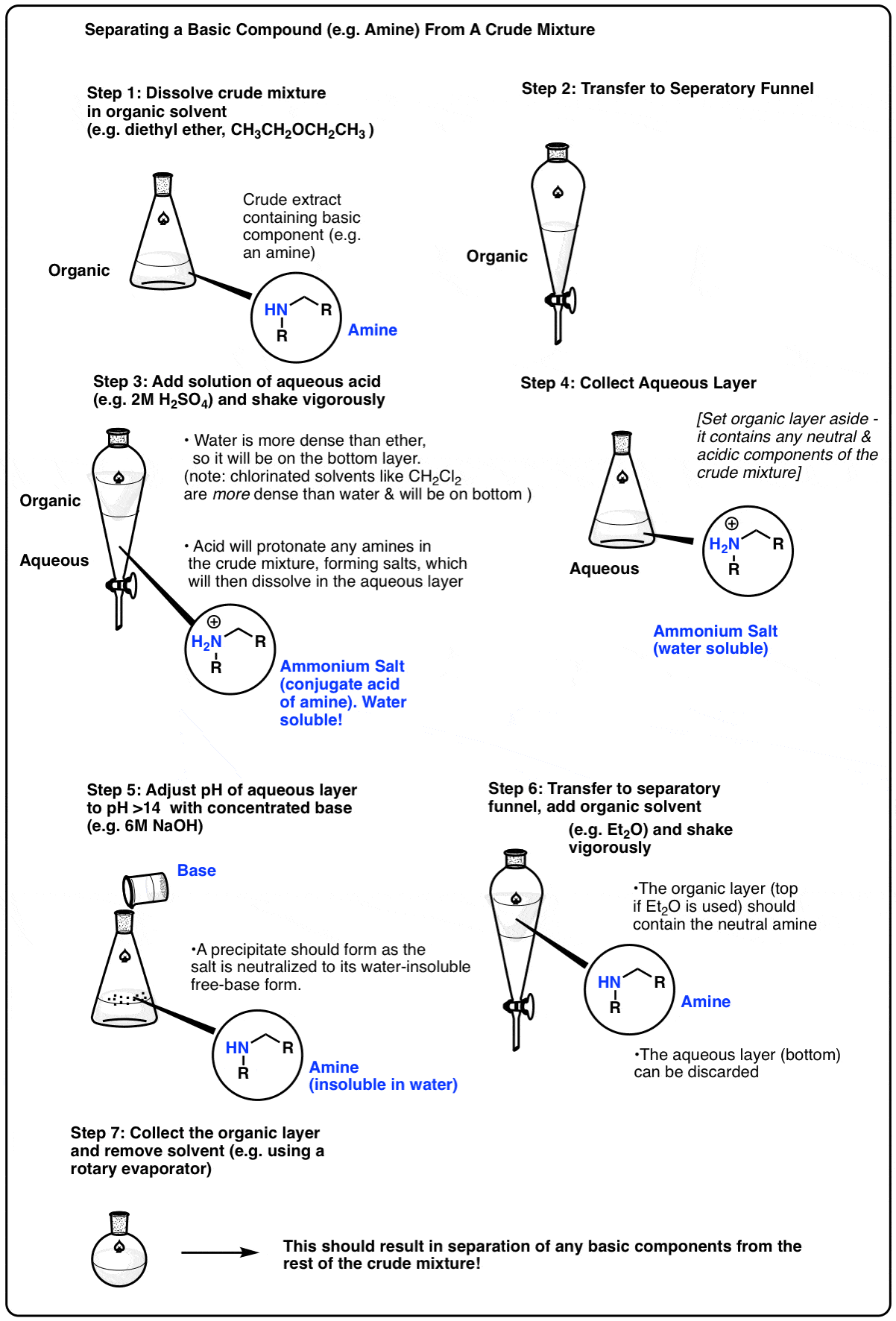
The separation of acidic and basic components from a crude mixture is often one of the first steps in separating the components of a crude natural product extract. It can save a lot of purification time if one knows beforehand that the desired compound of interest is acidic or basic.
2. Separation By Boiling Point Differences (Distillation)
Distillation is one of the most familiar purification methods: a flask containing the mixture is heated to boiling, and the vapor is condensed and collected. We monitor the temperature of the vapour with a thermometer and note the boiling point of each fraction. The composition of the vapor is a function of the boiling points of the components according to Raoult’s Law (ratios of vapour pressures).
An oil like lavender extract is composed of many different components with differing molecular weights and boiling points.In theory, one should be able to separate the components by distillation – right?
In practice, it’s not that easy using conventional lab equipment, unless there is good separation between boiling points (40° C is a decent rule of thumb). One can increase the separatory powers of distillation somewhat by employing a fractionating column, which results in more condensation-evaporation cycles and better separation.
[Scale is also an issue. With less than 1 mL of liquid it becomes tough to do an effective distillation because it’s difficult to heat the liquid evenly. On small scale, there are tools like the Kugelrohr which can help.]
What if you want to go beyond “conventional lab equipment” ? Well, you could build a refinery.

[Those tall towers are distillation columns]
After all, the point of a refinery is to take crude oil, separate the components by their boiling points and prepare the resulting fractional distillates for sale as gasoline, diesel fuel, kerosene, jet fuel, and others ( the residual crap that doesn’t distill of is what we call “asphalt”).
What we call “gasoline” is actually a mixture of over 200 distinct hydrocarbons from C4 to C12 that boils from 40-200 degrees C . Given a large enough distillation setup, these components can be further separated into their individual components such as pentane, hexane, heptane, and so on, which are used as fine chemicals.
3. Crystallization
We’re all familiar with crystals and the process of recrystallization, at least to some extent. But how can crystallization be used to obtain pure compounds from a crude mixture?
Here’s an everyday example with historical importance. Examination of the corks or bottom of certain wines will reveal tiny clear shards that resemble broken glass. These “wine diamonds” are in fact crystals of potassium ditartrate, which slowly crystallizes out of wine as the alcohol content increases. [Potassium ditartrate is fully soluble in (non-alcoholic) grape juice, but poorly soluble in ethanol]. Hence the crystals of bitartrate are separated out from the manifold other compounds present in wine.
You may be aware that tartrate crystals are also notable in that their study by Louis Pasteur gave rise to the discovery of optical isomerism.

A general method for recrystallization is as follows:
- Solvent survey. Examine a variety of potential solvents for the crude mixture. An ideal recrystallization solvent will dissolve the entire mixture at high temperature, but not at low temperature.
- Dissolve the crude mixture at high temperature, until no solids remain. It might be necessary to filter off any insoluble materials.
- Allow the mixture to cool undisturbed. Slower cooling tends to result in larger crystals.
If you are fortunate, you may be rewarded by the appearance of gleaming crystals at the bottom of the flask. If recrystallization is not occurring, it might help to scratch the flask to create nucleation sites for crystals to form. [One of the legends surrounding the origin of the name for “barbituric acid” was that it was so named because its crystallization was helped by scratching a chemists’ dandruff-laden beard (“barba” in Latin) over the crystallization dish. Chemists have never been known for their hygiene.]
If this is all jibber-jabber to you, here’s a video from MIT.
Another method is to add a small amount of co-solvent to the hot, dissolved crude mixture in which certain components are known to be insoluble. [Video]
Until only a few decades ago, recrystallization was one of the few methods of chemical purification and characterization available to chemists. If you couldn’t get crystals, forget it. [Note 1]. This could lead some chemists to desperate measures.
Crystallization is about as close to an ideal purification method as you can get. The products are generally highly pure (unlike the mixtures one can sometimes obtain with distillation), it is operationally simple, relatively cheap, and can be done on scales from a few milligrams up to hundreds of kilograms (and likely beyond).[Note 3]
Trying to purify that amount of material using chromatography (see below) is a nightmare.
The only problem is that not all compounds form crystals, and sometimes finding conditions that will selectively recrystallize one compound can be extremely time consuming. [Note 2]
Another important thing to note about crystallization is that the structure of unknown compounds can be determined by a technique called X-ray crystallography. This is how Dorothy Crowfoot Hodgkin determined the structure of Vitamin B12, work for which she was awarded the Nobel Prize in 1964. X-ray crystallography is the gold-standard of structure determination: with very few exceptions, if you can get a compound to crystallize, you can determine its structure.
Not all organic molecules can form crystals. Those that can often have fairly rigid structures with one or more rings, or are salts. [One of the reasons why early organic chemistry focused on steroids, heterocycles, and to a lesser extent alkaloids is that they are relatively easy to crystallize – very important in the days before modern chromatography techniques]. In the old days, one way to deal with the problem of molecules that wouldn’t crystallize was to make derivatives such as hydrazones or bisulphite adducts that were commonly crystalline or had characteristic melting points. We still force many undergraduate students to go through this rigamarole even though the usefulness of making derivatives has long passed.
If you’re interested in more information on crystallization, Brandon Findlay has a nice piece on it over at Chemtips.
We conclude this section with some crystal glamour shots.

4. Chromatography
For everything that can’t be easily purified by distillation or recrystallization there is column chromatography. Truth be told, for most bench chemists working on typical lab scale (from a few milligrams up to several grams of starting material) column chromatography is the go-to separation technique. Like the best answer to the Prisoner’s Dilemma, it might not be an ideal solution, but at least you usually have certainty about how much time it will cost you.
How does chromatography work? The best quick analogy I can think of is Velcro. Imagine a floor carpet made of Velcro “hooks”. Then imagine walking on it with your normal running shoes. No problem, right? Nice and smooth! Now imagine lining the bottom of your running shoes with Velcro “fuzz” and doing the same. You’ll walk much slower, and make annoying rip-rip noises, besides.
The “velcro” in our analogy represents interactions between the (polar) silica gel packed in the column (the “stationary phase”), containing free OH groups, and any polar groups dissolved in the solvent (the “mobile phase”) as they are passed through the column. The more polar groups the compound has, the more hydrogen-bonding interactions it will have with the polar silica gel, and the slower it will move down the column [like in our Velcro example].”Greasy” compounds – those with few polar groups – will move quickly down the column since they will interact very little with the silica gel [much like walking on a velcro carpet with “normal” shoes].
As this is turning into yet another ridiculously long post I am loath to go into too much specific detail on chromatography and am happy to refer you to the many great resources online (e.g. Youtube) where you can learn more about how to run a column. Again, Brandon Findlay from Chemtips has a whole series of posts devoted to it, which I recommend.
Here’s a rough outline of running a column.
- Using thin-layer chromatography (TLC) plates, dilute samples of the crude mixture are “spotted” and placed in various solvent mixtures in order to find conditions where good separation can occur.
- Based on the amount of material to be separated, an appropriate size of column is chosen and packed with silica gel and a starting solvent (usually hexane).
- The crude material is loaded on top of the column dissolved in a minimum amount of non polar solvent.
- Eluent (the solvent system “mobile phase” determined during the TLC experiments) is added to the top of the column, and pressure is applied.
- The solvent is run through the column and collected in small tubes. Appearance of the various products is monitored by TLC (and UV lamps, if the material absorbs in the UV – many do!).
- After determining which tubes contain which compounds (by TLC) the fractions are collected and concentrated, and then examined by a spectroscopy technique such as NMR (more on that in future posts).
Once you have a few columns under your belt, you can do most simple purifications in under an hour on typical scale (50-100 mg is common for test reactions). As the amount of material climbs into the >10g scale, however, that’s when we start looking for other techniques – running the column and removing all the solvent can take the better part of a day on large scale!
Final note. On small scale (less than 50 mg) it can also be useful to attempt preparative thin layer chromatography, which involves using a much larger plate. After development, the different fractions can be visualized by UV (if the compound absorbs in the UV), the silica scratched off, and the compounds removed from the silica gel with a solvent like ethyl acetate. Here’s a pic.
5. Gas Chromatography (GC) and High Performance Liquid Chromatography (HPLC)
OK, so maybe you don’t have a kilogram of crude material to purify. Maybe you don’t even have a gram, or 10 milligrams. Maybe you have a milligram or less (not uncommon for some natural product isolations!). Unfortunately, all of the techniques mentioned above are out. Thankfully, there’s still an option. GC and HPLC to the rescue!
GC and HPLC require expensive instruments and a significant time investment in learning how to use them, and are definitely not available in all labs. However, for the purpose of giving an overview of all the compounds present in a mixture, they are unmatched. No other technique can deliver a result like that analysis of lavender oil (and its 36 compounds) I put at the top of this post. Or this analysis of a mix of terpenes by HPLC on 10 microlitres of material. Look at that separation [source].
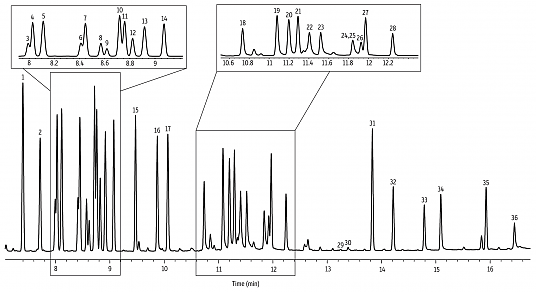
Without getting into much detail, these are forms of chromatography that follow the same principles as column chromatography (above) except that the “column” is much smaller in diameter and run at significantly higher pressures. As the name suggests, the “solvent” (mobile phase) is an inert gas in the case of GC and liquid (often hexane / acetonitrile) in the case of HPLC.
Besides their great powers of separation, a further advantage of HPLC and GC is the fact that analyses can be done on an astonishingly small amount of sample. For example, the sample above was run on two micrograms. Your sweaty fingerprints weigh more than that!
If pure samples are desired it’s possible to use GC and HPLC to isolate enough material to allow for full characterization of a compound using larger columns. This is referred to as “preparative” GC and HPLC and is particularly valuable for small, valuable samples. About 1 mg of material is sufficient to be able to fully characterize an unknown compound using our modern spectroscopic techniques (mostly NMR).
6. Conclusion – Overview of Purification Techniques
This has been a long post. The point has been to give an overview of the key techniques used for purifying crude mixtures: 1) chemical properties, 2) distillation, 3) crystallization, 4) chromatography, 5) GC/HPLC.
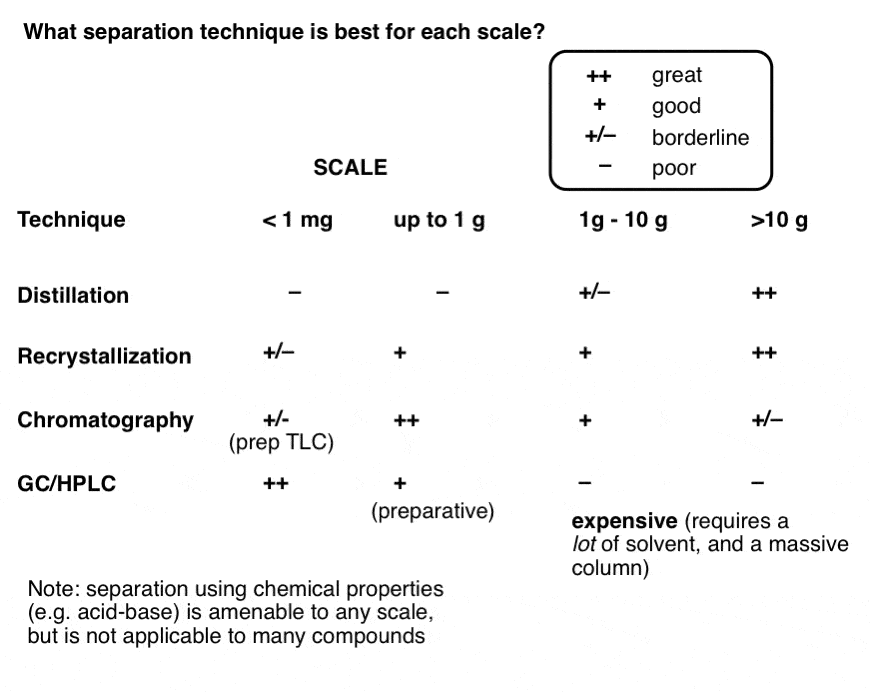
Here’s a handy table summarizing the advantages and disadvantages of each for each scale. Opinions are my own. Disagreements welcome in the comments.
In the next post, we’ll move beyond this overview and start to ask the question: how do you characterize the structure of a pure, unknown compound?
Notes
Note 1. An emeritus professor I worked with once told me that an entire PhD in late 19th century Germany might involve heating various compounds for days with concentrated acid, followed by various attempts at recrystallizing a product from the resulting soup. An entire Ph.D could consist of characterizing the products of one reaction.
Note 2. The same professor told a tale (possibly aprocyphal) of a graduate student who was so frustrated with the inability of his compound to crystallize after countless attempts that he took a piss in his recrystallization dish. Miraculously, crystals of his desired compound then appeared. Apparently, it was reported in the experimental as “co-crystallized with uric acid”.
There is also a chromatography technique which is actually older than HPLC but still relatively unknown despite it does not use a column, only solvents so scaling up from laboratory scale to industrial scale is almost linear. It is called Centrifugal Partition Chromatography (or CPC in short) and it uses a biphasic solvent system which can consist of multiple solvents. One of the two phases will be the stationary phase, and the other will be the moving phase (in this case deciding between the more dense or less dense phase to be the stationary one is like deciding between normal and reverse phase column). The stationary phase is immobilized by the spinning of a rotor (centrifugal force), so it will act similarly to a solid phase, while the mobile phase is percolated through it with high pressure. The drive of the separation is the difference between the partition coefficient of each compounds.