Enols and Enolates
Kinetic Versus Thermodynamic Enolates
Last updated: May 28th, 2026 |
Kinetic versus Thermodynamic Enolates of Ketones
Enolates have a lot in common with alkenes. They are flat and have a C-C pi bond.
- Zaitsev’s rule reminds us that alkene stability increases with increasing number of carbons directly attached to the alkene (i.e. “more substituted” alkenes are more stable).
- When enolates are formed with strong, non-bulky bases (e.g. NaOH or NaOCH3) the tendency is for the more substituted enolate to form. When the enolate reacts with electrophiles, this means that the electrophile will add to the more substituted alpha carbon.
- We can overcome this tendency of enolates by using a strong, bulky base such as LDA (lithium di-isopropylamide) which reacts with the less sterically hindered proton faster than it does with the more sterically hindered proton.
- This “less substituted” enolate is called the “kinetic enolate“.
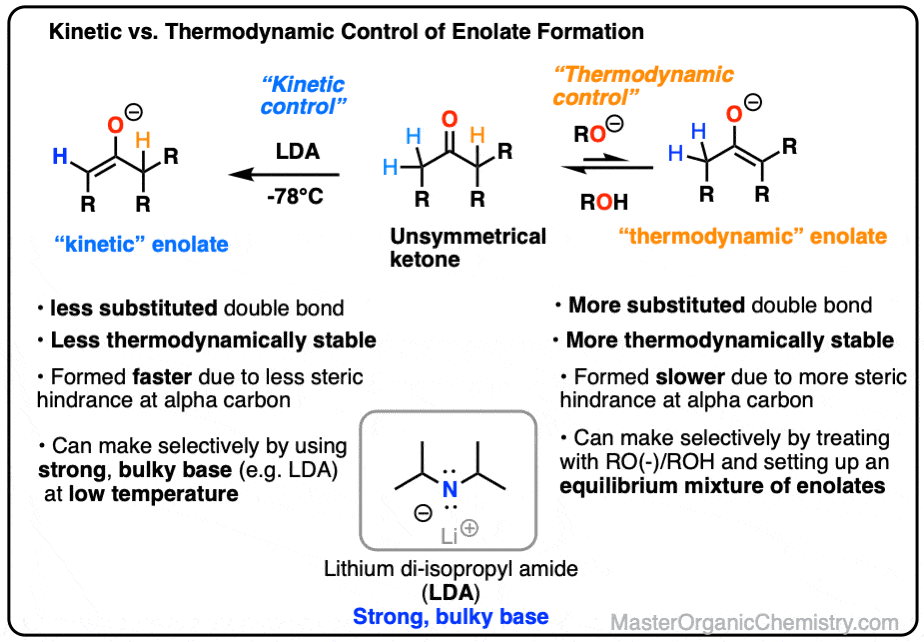
Table of Contents
- Thermodynamic Enolate Formation
- Reactions of Thermodynamic Enolates
- Selective Formation of “Kinetic” Enolates
- Reactions of Kinetic Enolates
- The Further Adventures of LDA: Nitrile and Amide Enolates
- Summary
- Notes
- Quiz Yourself!
- (Advanced) References and Further Reading
1. Thermodynamic Enolate Formation
If you aren’t familiar with enolates and need more background, go back and read the previous post on enolates first. [See post: Enolates]
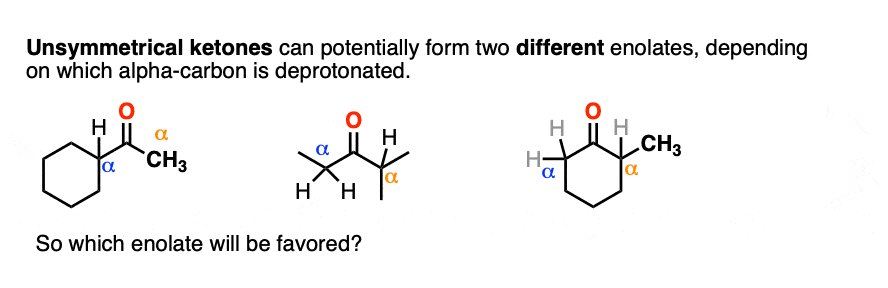
Many ketones are capable of forming two different enolates, depending on which alpha-carbon is deprotonated.
The question is, which one will be favored?
Generally enolates are flat and can be thought of as similar to alkenes, even if they tend to react with electrophiles like they are carbanions.
Going back to Zaitsev’s rule, we’ve seen many examples where alkenes increase in stability as the number of H atoms directly attached to the ring decreases. (or conversely, as the number of attached carbons increases)
In other words, the more substituted the alkene, the more stable it is. [Note 1]
This also applies to enolates. The fewer C-H bonds there are on the alkene, the more thermodynamically stable it is.
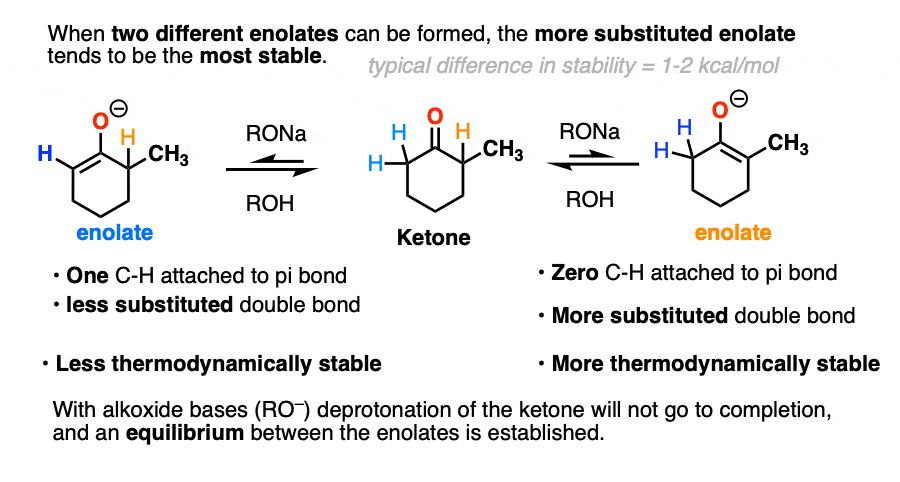
Ketones can undergo deprotonation with strong bases like alkoxides RO(-) to give enolates.
Alkoxides are not as basic as ketone enolates, so the acid-base equilibrium tends to favor the starting ketone. However, they are still strong enough bases to set up an equilibrium between the starting ketone and the two different enolates.
The position of that equilibrium will favor the most thermodynamically stable enolate (i.e. the most substituted), even though it is slightly slower to form due to the fact that the C-H bond is more sterically hindered.
For that reason we call the more substituted enolate the thermodynamic enolate because of its greater stability.
We also say that formation of this enolate is under thermodynamic control.
The equilibrium ratio of enolates will depend on the difference in energy between their heats of formation (which is typically 1-2 kcal/mol). As a rough rule of thumb, 4:1 is a good ballpark number but it can vary considerably. [Note 2]
2. Reactions of Thermodynamic Enolates
These “thermodynamic” enolates can act as nucleophiles in various reactions.
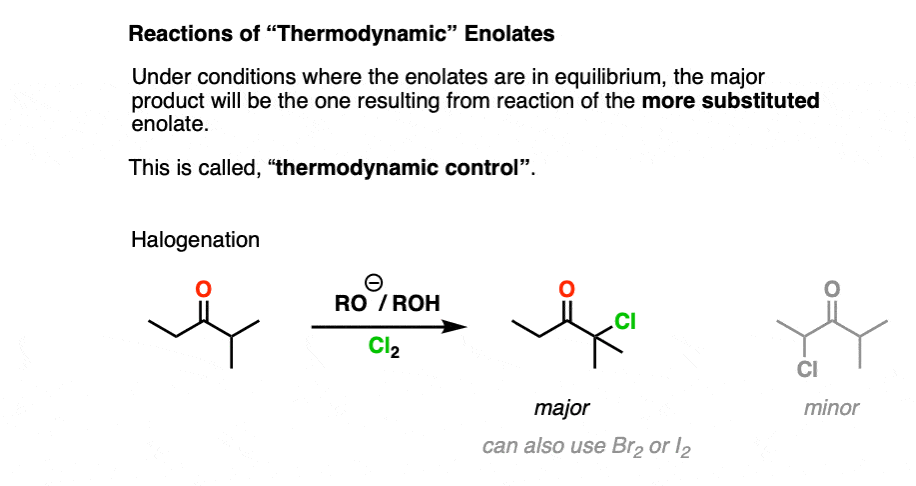
Any time we form an enolate under thermodynamic control, we should expect that the major product will arise from the reaction of the more substituted enolate with the electrophile, such as in this halogenation reaction. [Note 3]
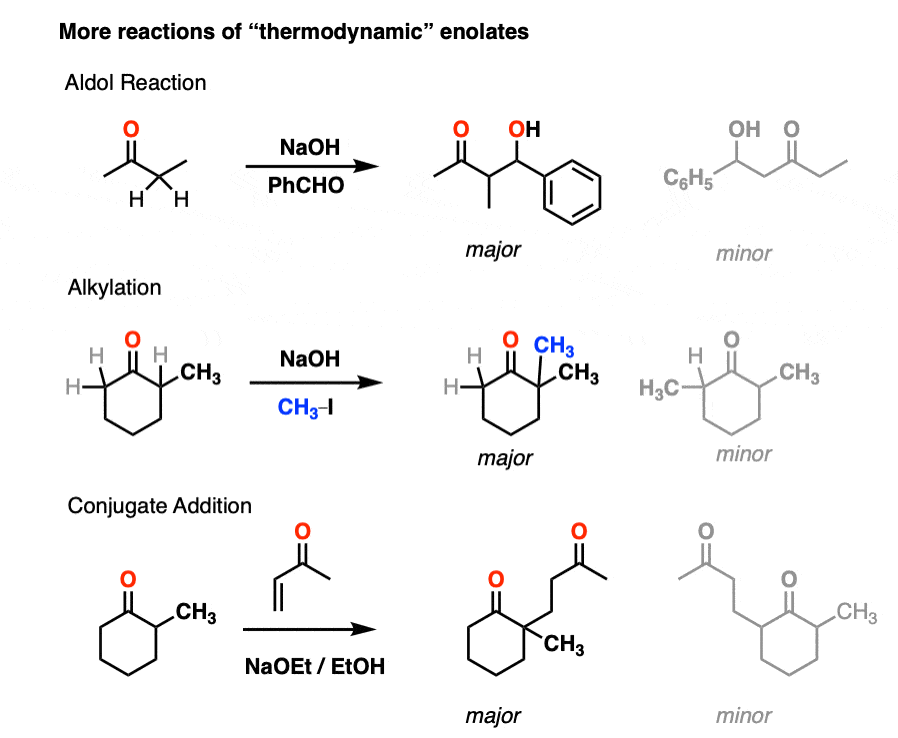
Similarly, this will also be the case in these examples of the Aldol reaction, enolate alkylation, and conjugate addition. [Note 4]
You may ask, “is that it?”
Are we doomed by this thermodynamic preference of the enolate to never be able to form the other less substituted enolate, just because it screams out, “Thermodynamically, I don’t wanna!“.
No! There’s a workaround!
3. Selective Formation of Kinetic Enolates
There are lots of times we might want the less-substituted enolate. So here is a strategy for how to go about making it.
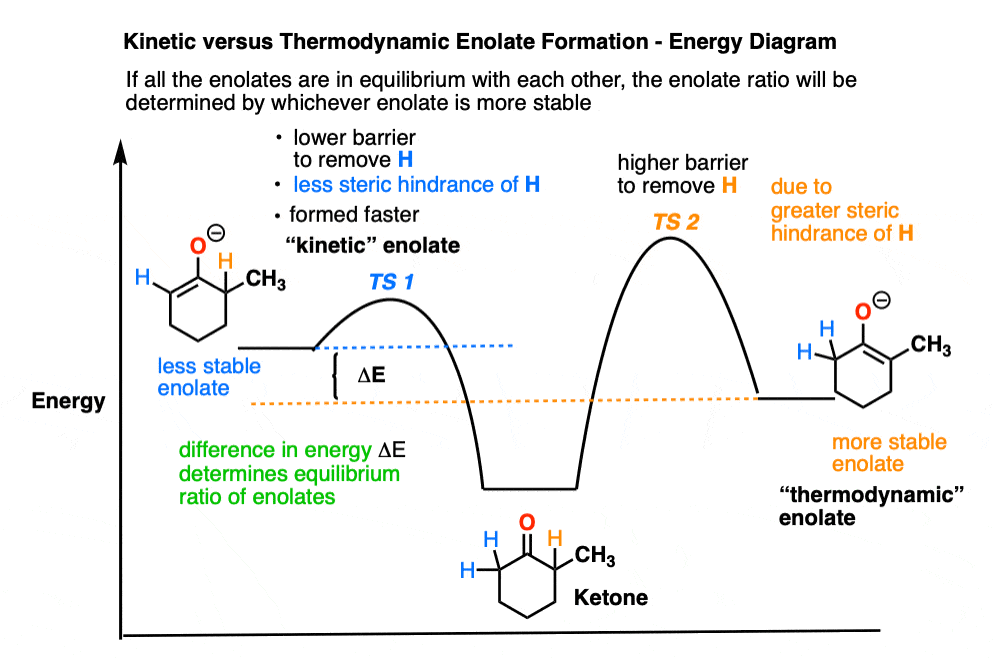
The first thing to note is that the hydrogen on the more-substituted side is slightly more difficult to access due to the presence of the extra alkyl group.
That’s why there is a higher energy barrier for deprotonation in the energy diagram (above).
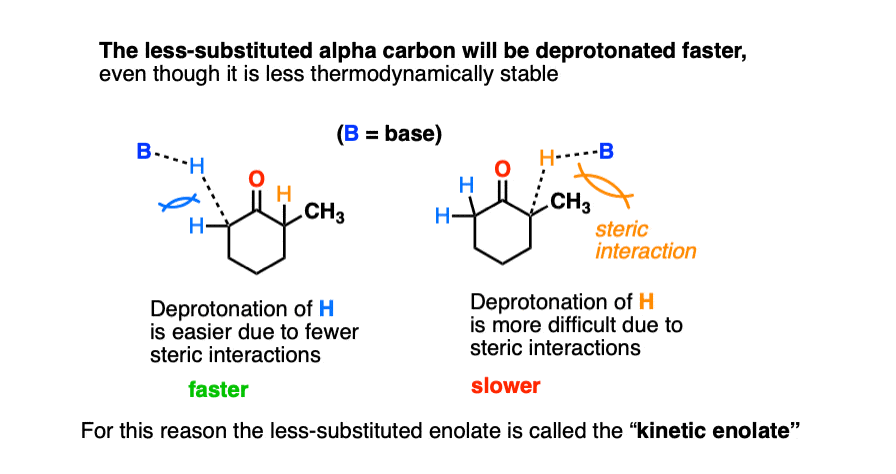
So what if we were to use a base that is extremely sterically hindered?
In that case the reaction with the more sterically hindered proton should be very slow, and reaction with the less sterically-hindered protons on the other side should be fast.
A good choice for this is the strong bulky base, lithium di-isopropyl amide (LDA).
LDA has two big and bulky isopropyl groups flanking a very basic amide base. The pKa of the conjugate acid is 38, so deprotonating a ketone alpha-carbon (pKa 16-18) is no problem for LDA.
Also, unlike alkoxide bases RO(-), deprotonation goes to completion. So long as an excess of base is used, there is no equilibrium between the different enolates. [Note 5]
LDA is so strong that deprotonation can happen at extremely low temperature. This helps us because we can use low temperatures to slow down that undesirable acid-base reaction even more.
So when our ketone is treated with LDA at low temperature we get preferential formation of the less-substituted enolate. We call this, “kinetic control”.
Note that there is nothing magic about -78°C. That just happens to be the temperature of the convenient (and cheap) dry ice-acetone cooling bath.
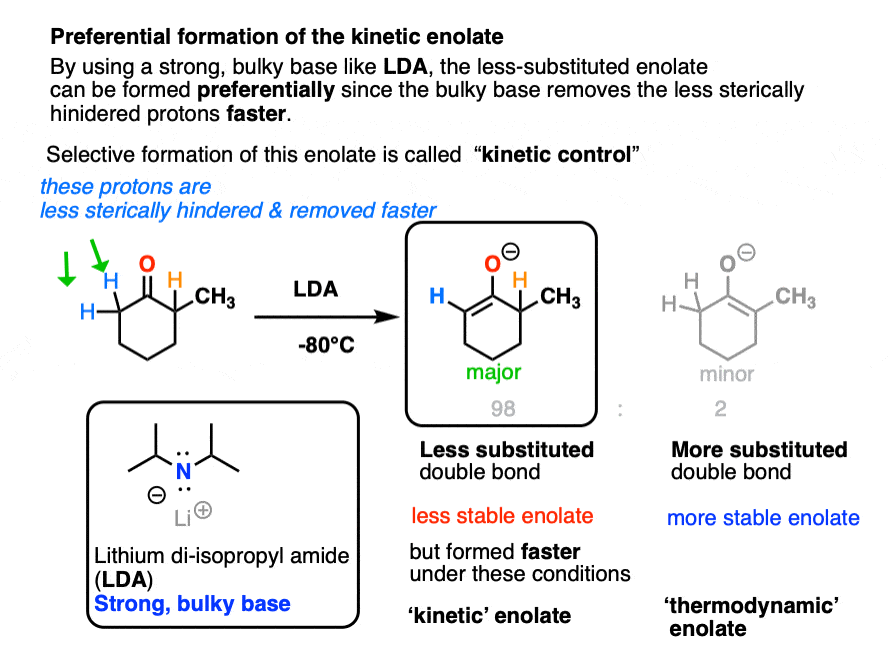
We call the less substituted enolate the “kinetic enolate” because we are depending on the difference in reaction rates (“chemical kinetics” remember?) to give us selectivity.
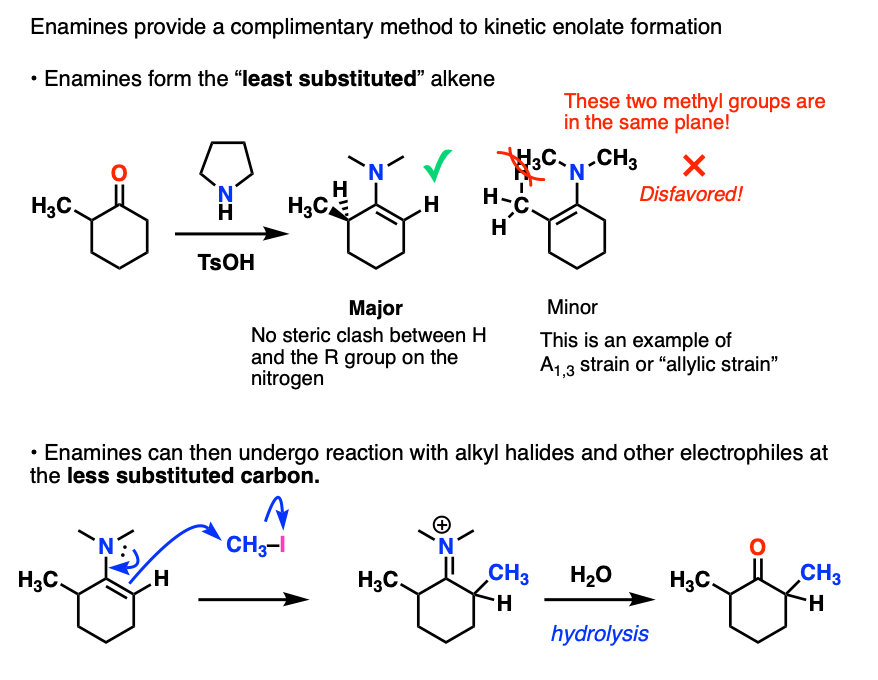
Note that enamines can also be used for performing reactions at the less-substituted alpha carbon. [Note 6]
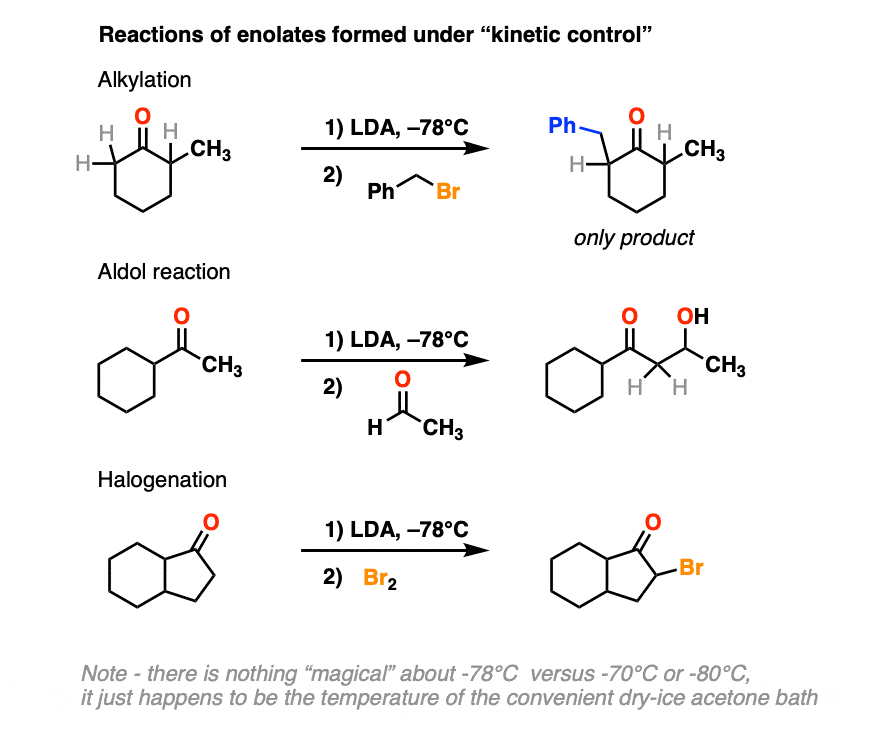
4. Reactions of Kinetic Enolates
Kinetic enolates can be used for the same reactions of enolates we’ve seen previously, such as alkylation, the Aldol reaction, and halogenation.
In each case we are forming our new bond at the less substituted alpha carbon of the ketone.
5. The Further Adventures of LDA
There’s another advantage to using LDA. Since LDA is such a strong base, we can use it form some of the less-accessible enolates of carboxylic acid derivatives such as esters, amides, and nitriles.
These enolates can perform the same types of reactions as those we’ve seen above, such as alkylation and conjugate addition.
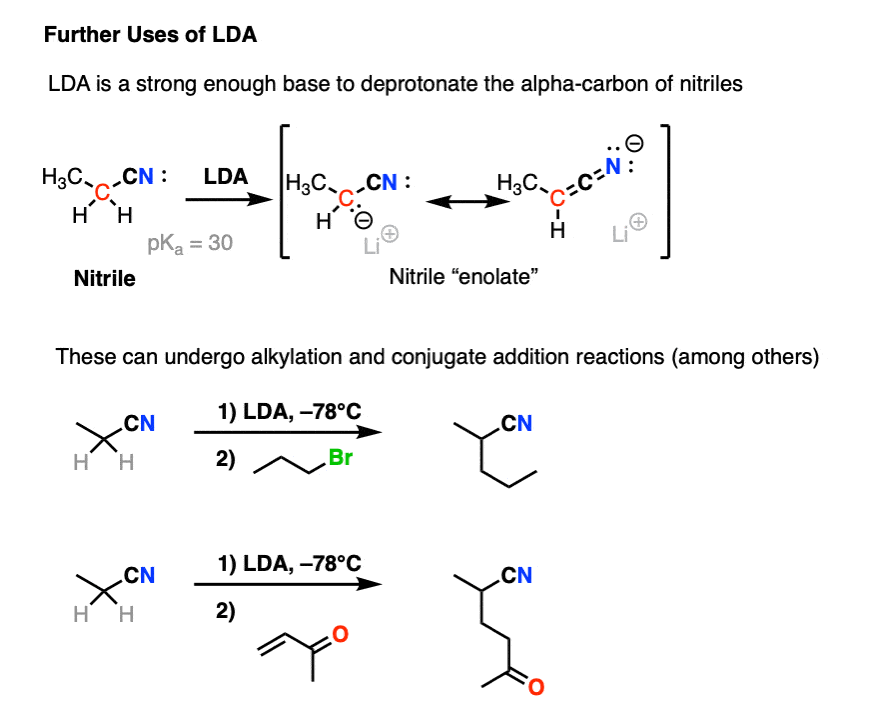
In short, LDA is an extremely useful strong base that can form just about any enolate you need, provided that the C-H bond isn’t sterically hindered. [Note 7]
6. Summary
So what have we learned?
- Unsymmetrical ketones are capable of forming two different enolates
- When alkoxide bases are used (RO-/ROH) there is incomplete deprotonation. As a result, an equilibrium exists between the starting ketone and the two possible enolates
- Equilibrium generally favors the more substituted enolate since you can think of it as a more substituted double bond. For this reason the more substituted enolate is called the thermodynamic enolate.
- By using a strong bulky base, the rate of deprotonation at the more substituted enolate can be slowed to the point where only the less-substituted enolate forms. This is referred to as the kinetic enolate.
- LDA (lithium diisopropyl amide) is a strong, bulky base that can be used for deprotonation of ketones, esters, amides, nitriles, and more.
Notes
Note 1. A rough rule of thumb is that each C-H you replace with a C-C gets you an additional 1 kcal/mol of stability. This might not sound like a lot, but even 1 kcal/mol is enough to give you about 80:20 equilibrium ratio.
Note 2. “it can vary considerably”. A typical ratio of thermodynamic:kinetic enolates under “thermodynamic conditions” (NaOR/ROH) is about 4:1 but it can be significantly larger than that. The identity of the alkali metal counter-ion can have a huge role. Page 2 of this article provides a great overview on forming thermodynamic enolates.
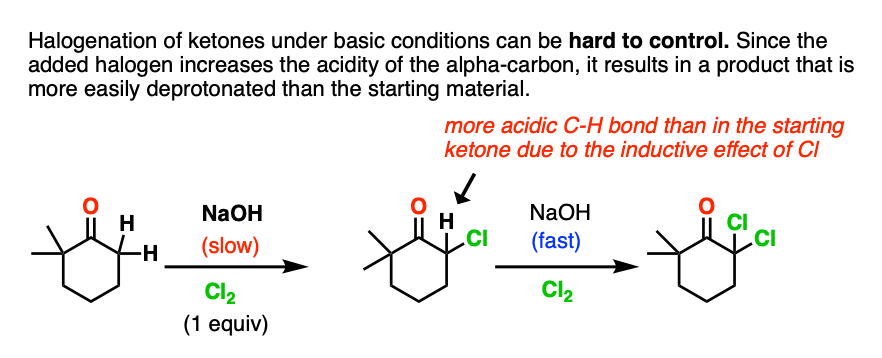
Note 3. .This reaction can be hard to control! One problem with halogenation of ketones under thermodynamic conditions (RO(-) / ROH) is that the halogenation product is more acidic than the starting ketone. This can easily lead to multiple halogenations, as in the Haloform reaction.
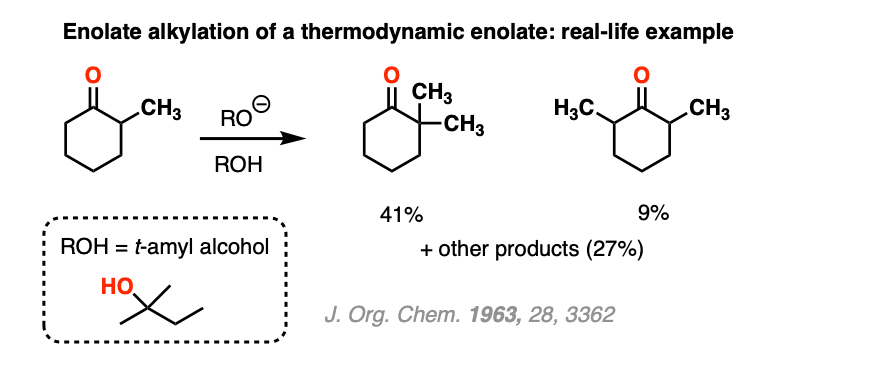
Note 4. For more on alkylation of ketones under thermodynamic conditions, see this classic study. It turns out the reaction of the ketone with NaOR/CH3I does indeed put CH3 on the more substituted enolate (in 41% yield), but there are lots of byproducts due to over-alkylation. For the purposes of our course, this is why procedures like the acetoacetic ester synthesis are preferred for enolate alkylation.
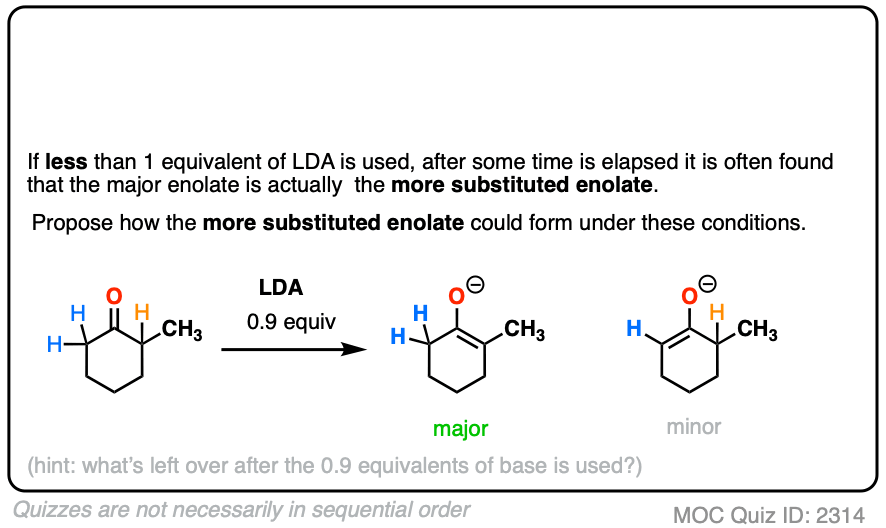
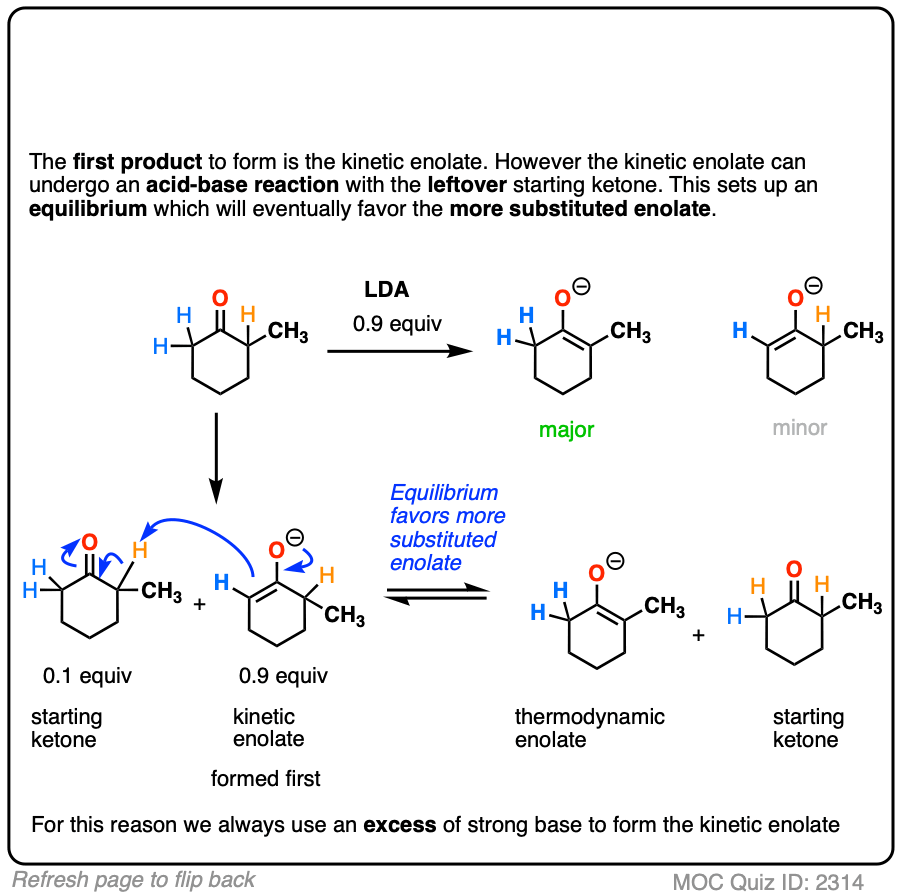
Note 5. “No equilibration between the enolates”. If less than 1 equivalent of strong base is used, it’s possible to end up with the thermodynamic enolate after some time has elapsed.
 Click to Flip
Click to Flip
Note 6. An alternative to kinetic enolate formation is to form the enamine which tends to favor the less-substituted alkene.
Note 7. LDA isn’t particularly useful for the deprotonation of aldehydes. It tends to perform nucleophilic addition to them instead.
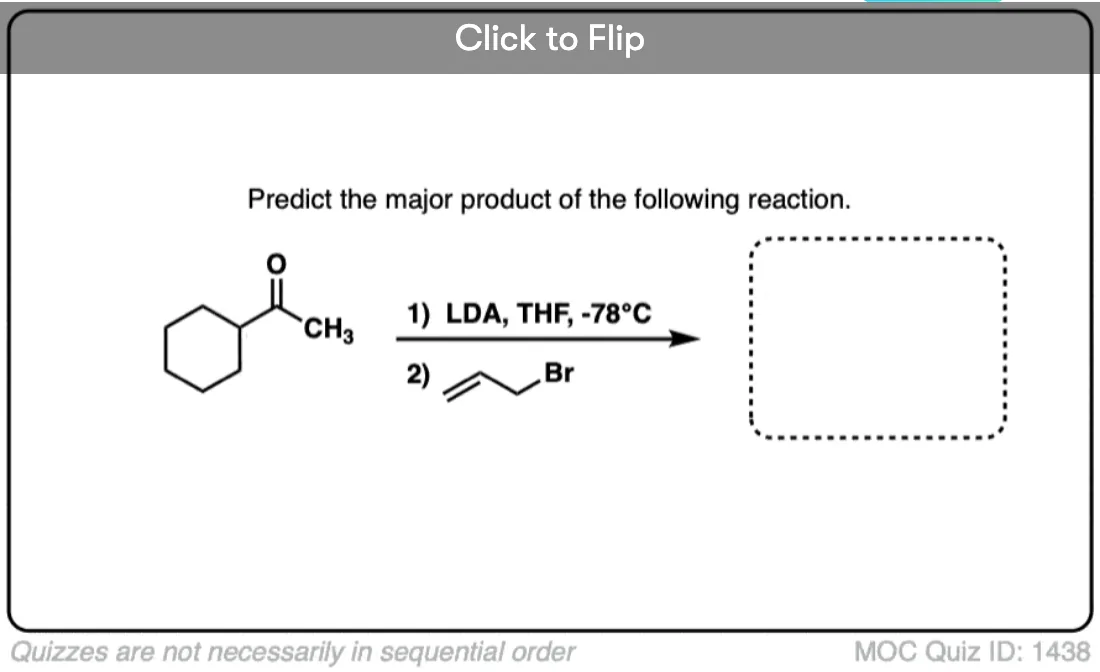
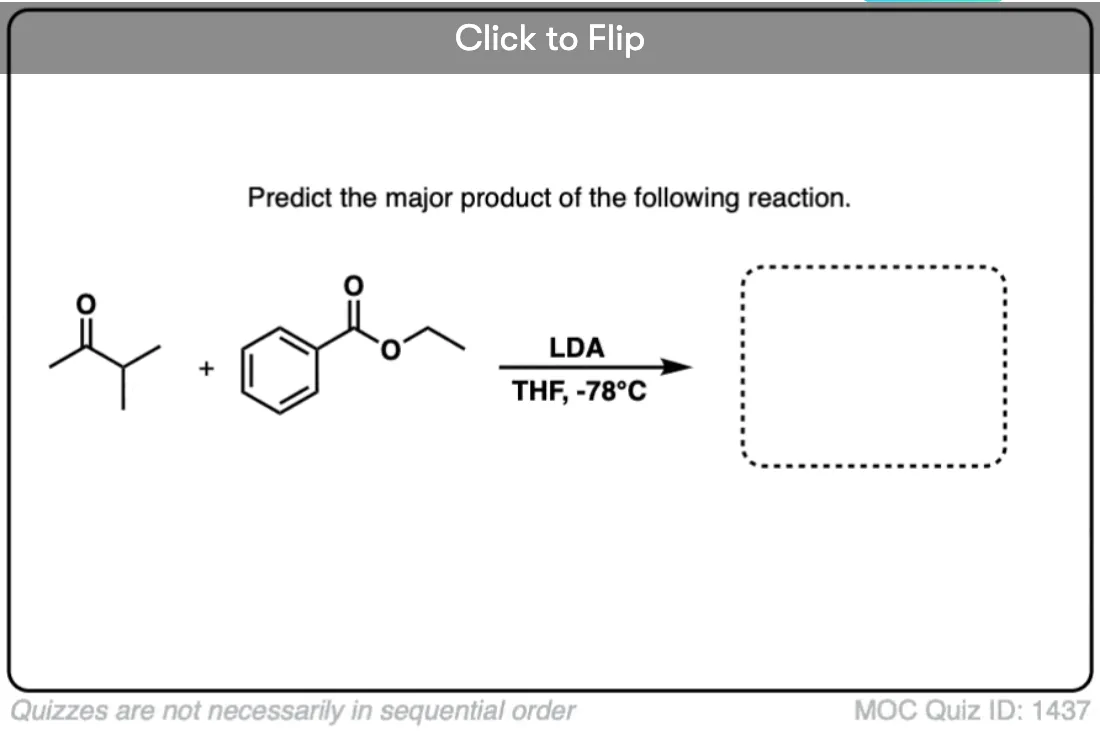
Quiz Yourself!
Become a MOC member to see the clickable quiz with answers on the back.
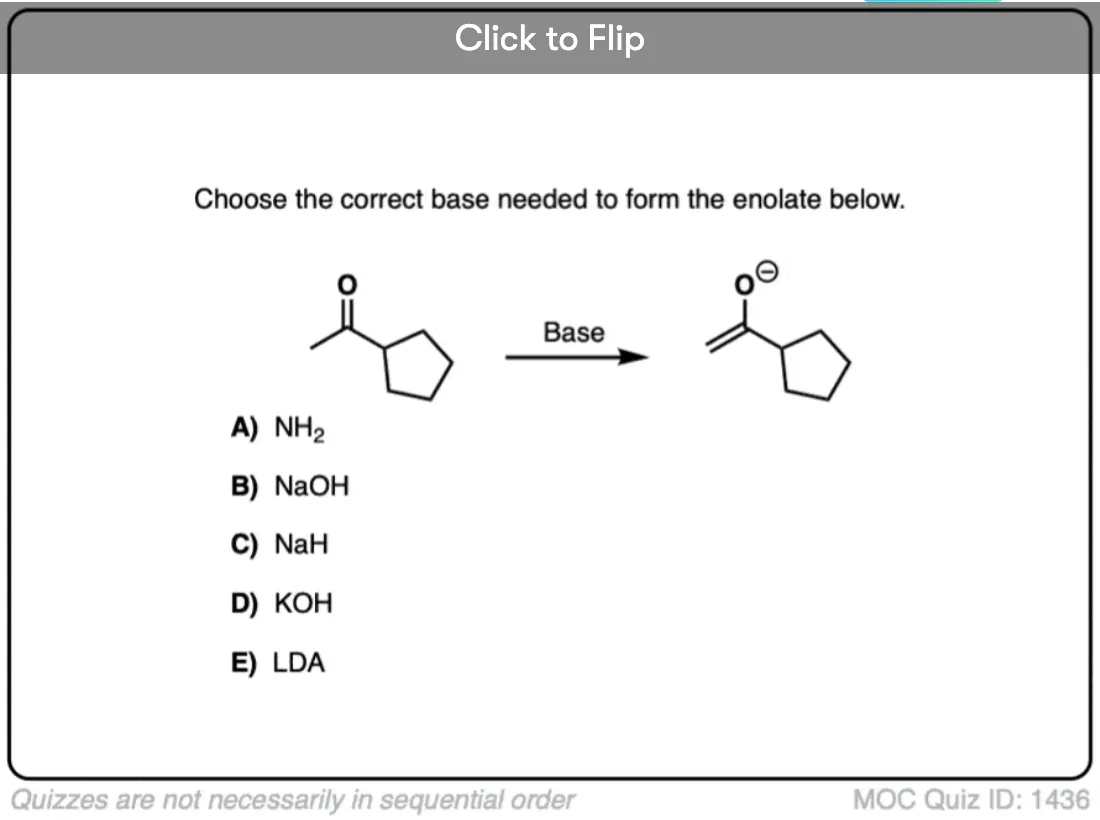
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
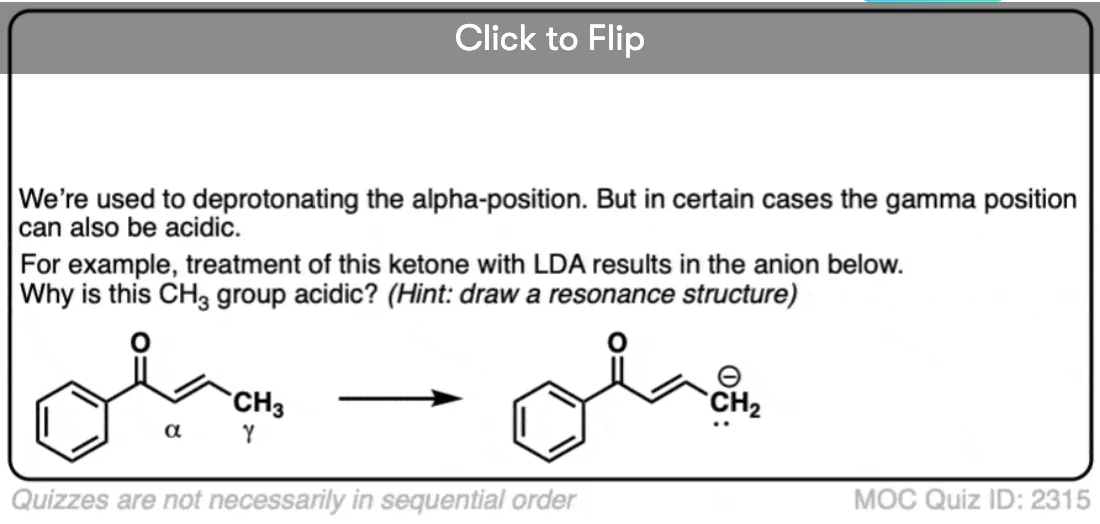
Become a MOC member to see the clickable quiz with answers on the back.
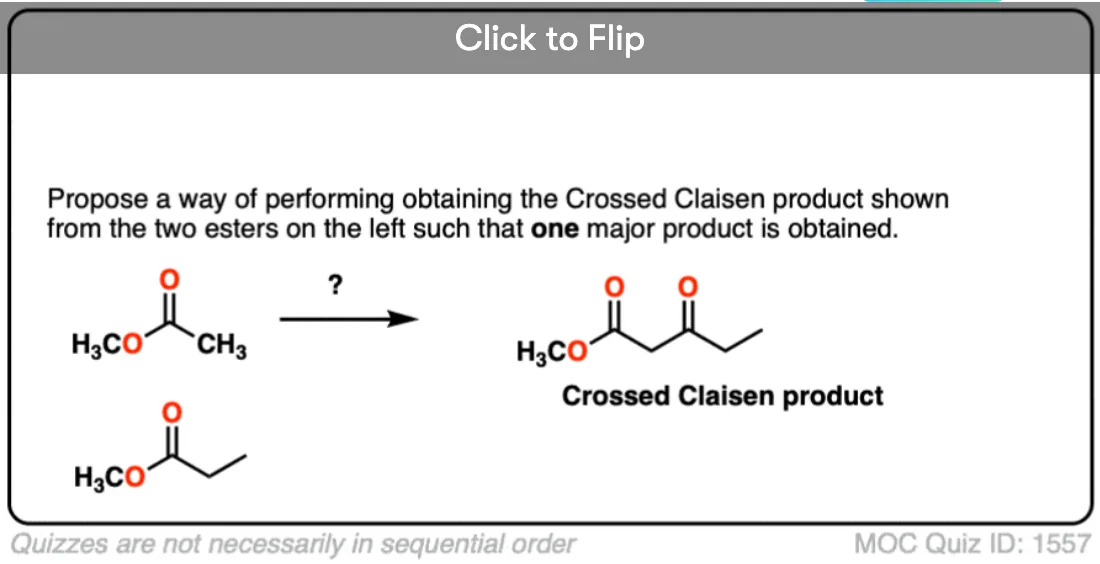
Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading
References
- THE FORMATION AND ALKYLATION OF SPECIFIC ENOLATE ANIONS FROM AN UNSYMMETRICAL KETONE: 2-BENZYL-2-METHYLCYCLOHEXANONE AND 2-BENZYL-6-METHYLCYCLOHEXANONE
Martin Gall and Herbert O. House
Org. Synth. 1972, 52, 39
DOI: 10.15227/orgsyn.052.0039
This procedure from Organic Syntheses has detailed experimental conditions for regioselective alkylation of an unsymmetrical ketone through kinetic and thermodynamic enolate formation conditions. - THE α-ALKYLATION OF ENOLATES FROM THE LITHIUM-AMMONIA REDUCTION OF α,β-UNSATURATED KETONES
Gilbert Stork, Perry Rosen, and Norman L. Goldman
Journal of the American Chemical Society 1961, 83 (13), 2965-2966
DOI: 10.1021/ja01474a051
This paper has one of the first descriptions of kinetic enolate formation in the literature – “The success of the trapping of the enolate ion IV depends on the alkylation reaction being faster than equilibration of the initially produced enolate IV to the more stable II via proton transfer with some initially formed neutral alkylated ketone.” - Tetrahedron report number 25: Ketone enolates: regiospecific preparation and synthetic uses
Jean d’Angelo
Tetrahedron 1976, 32 (24), 2979-2990
DOI: 10.1016/0040-4020(76)80156-1
This review covers various methods for enolate formation, and has data on the composition of various ketone-enolate mixtures formed under kinetic and thermodynamic conditions - Thermodynamic and Kinetic Controlled Enolates: A Project for a Problem-Oriented Laboratory Course
Augustine Silveira Jr., Michael A. Knopp, and Jhong Kim
Journal of Chemical Education 1998, 75 (1), 78
DOI: 1021/ed075p78
A paper from J. Chem. Ed. that covers how to demonstrate the concepts of kinetic and thermodynamic enolates in an undergraduate laboratory session. - The Chemistry of Carbanions. V. The Enolates Derived from Unsymmetrical Ketones
Herbert O. House and Vera Kramar
The Journal of Organic Chemistry 1963, 28 (12), 3362-3379
DOI: 10.1021/jo01047a022 - The Chemistry of Carbanions. IX. The Potassium and Lithium Enolates Derived from Cyclic Ketones
Herbert O. House and Barry M. Trost
The Journal of Organic Chemistry 1965 30 (5), 1341-1348
DOI: 10.1021/jo01016a001 - Kinetic Enolate Formation by Lithium Arylamide: Effects of Basicity on Selectivity
Linfeng Xie, Keith Vanlandeghem, Kurt M. Isenberger, and Carolyn Bernier
The Journal of Organic Chemistry 2003, 68 (2), 641-643
DOI: 10.1021/jo0263465
This paper is on the topic of enolate formation and examines how the stereochemistry of the enolate formed varies depending on the nature of the Li-amide base. - A stereoselective total synthesis of (±)-gymnomitrol
Steven C. Welch and Suthep Chayabunjonglerd
Journal of the American Chemical Society 1979, 101 (22), 6768-6769
DOI: 1021/ja00516a057
The first step in this total synthesis is the alkylation of the Li enolate of 2-methylcyclohexanone, which is made by deprotonation by LDA at -78 °C, followed by equilibration to the thermodynamically more stable substituted enolate anion at room temperature for 4-5 hours. - Pseudoephedrine as a Practical Chiral Auxiliary for the Synthesis of Highly Enantiomerically Enriched Carboxylic Acids, Alcohols, Aldehydes, and Ketones
Andrew G. Myers, Bryant H. Yang, Hou Chen, Lydia McKinstry, David J. Kopecky, and James L. Gleason
Journal of the American Chemical Society 1997, 119 (28), 6496-6511
DOI: 1021/ja970402f
The enolates derived from N-acylation of pseudoephedrine can be selectively facially alkylated. It is easier to induce diastereoselectivity in a reaction than enantioselectivity, and one way to do that is to use a chiral auxiliary – a chiral scaffold that you attach to the substrate and then remove after you’ve alkylated it. This is a paper that demonstrates practical chemistry – at the time, pseudoephedrine was easier to obtain than it is now, and Prof. Myers (now at Harvard) has extensively documented the reaction and the procedures in order to make it reproducible, robust, and reliable. - Asymmetric alkylation reactions of chiral imide enolates. A practical approach to the enantioselective synthesis of .alpha.-substituted carboxylic acid derivatives
D. A. Evans, M. D. Ennis, and D. J. Mathre
Journal of the American Chemical Society 1982, 104 (6), 1737-1739
DOI: 10.1021/ja00370a050
This is a landmark paper, in which Prof. Evans pioneered the use of chiral auxiliaries and showed how they could be used for asymmetric aldol reactions. The auxiliary used here is a chiral oxazolidinone, and this chemistry is known as the “Evans Asymmetric Aldol” or the “Evans chiral auxiliary”.
I was taught that using LDA at -78 °C will give the kinetic enolate while using it at a higher temperature will give the thermodynamic enolate. Is that not true?
Wouldn’t using an alkoxide base for α-alkylation reactions give a large amount of SN2 products where the alkoxide reacts with the alkyl halide?
Hi, sorry for late reply. Thanks for asking!
1) Using an *excess* of LDA (e.g. 1.1 equivalents) at low temperatures will give the kinetic enolate.
The reason for low temperatures is twofold. First of all, at higher temperatures (e.g. room temp) there is a side reaction where LDA starts to deprotonate THF, leading to its decomposition. Secondly, for any process where you’re trying to get good selectivity, lowering the temperature will give you a little extra selectivity “for free”. [For more on this, see: https://www.masterorganicchemistry.com/2023/08/02/equilibrium-constant-delta-g-calculations-organic-chemistry/#five section 5]
At higher temperatures with an excess of LDA, you don’t get the kinetic enolate. You decompose THF.
2) To get the thermodynamic enolate, one typically uses a *deficit* (e.g. 0.95 molar equivalents) of a strong base (let’s say LDA) and let it warm slightly (e.g. -20 degrees C). This results in an equilibrium between the enolate and the ketone, and eventually the ratio of enolates will reflect the thermodynamic ratio.
3) For the purposes of this article, I fib (as do many textbooks) and show a very simple method for making a thermodynamic enolate (alkoxide) which is very different from the “industry standard” way of making a thermodynamic enolate. I would also not recommend it in the lab for alkylation reactions. You’ve hit on one reason why (SN2 of alkoxides with the alkyl halide, so we must use an excess of both base and alkyl halide). Also the selectivity is not great.
I hope this helps. Thanks for the questions!