Reactions of Aromatic Molecules
EAS Reactions (3: Friedel-Crafts Acylation and Friedel-Crafts Alkylation
Last updated: May 29th, 2026 |
Friedel Crafts Alkylation and Acylation
- Aromatic rings will form C-C bonds when treated with alkyl or acyl halides in the presence of a strong Lewis acid (e.g. AlCl3). These are known as Friedel Crafts reactions and are examples of electrophilic aromatic substitution reactions.
- The Lewis acid coordinates to a lone pair on the halogen, making the halogen a better leaving group.
- In Friedel-Crafts alkylation, an alkyl halide treated with a Lewis acid results in a carbocation electrophile (or a species very similar to a carbocation) that is then attacked by the aromatic ring.
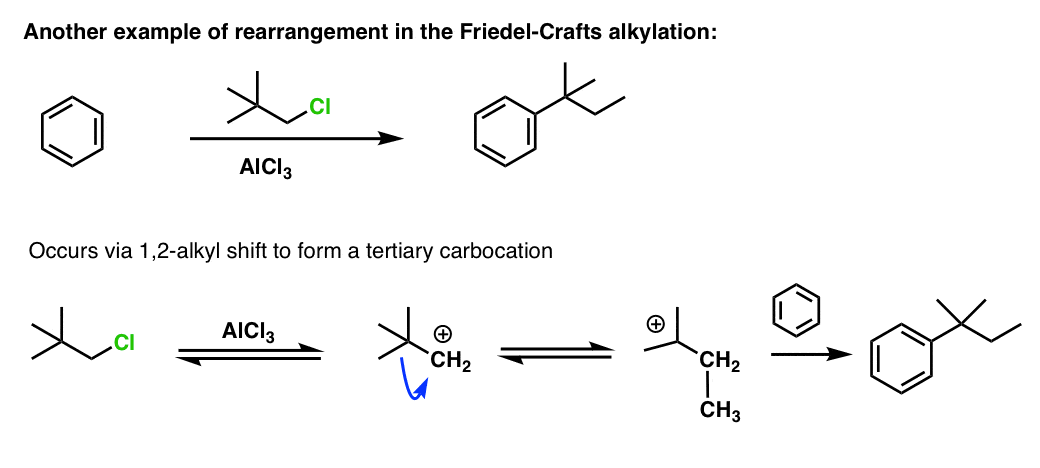
- Carbocation rearrangements can occur if they result in a more stable carbocation!
- Friedel-Crafts acylation is performed with acyl halides and Lewis acids. No rearrangements are observed.
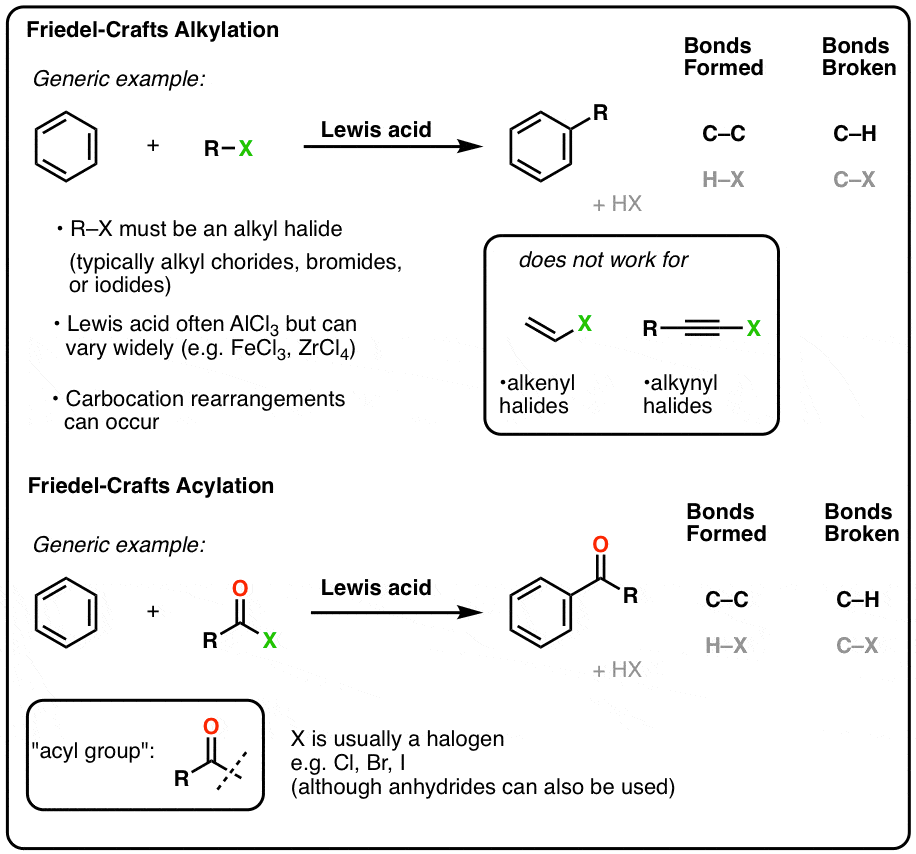
Table of Contents
- Quick Recap on Electrophilic Aromatic Substitution: Onward To Carbon-Carbon Bond Forming Reactions!
- Friedel-Crafts Alkylation Of Aromatic Rings
- The Role of The Lewis Acid In Friedel-Crafts Alkylation Is To Activate The Alkyl Halide
- The Key C-C Bond Forming Step Is Attack Of The Aromatic Ring Upon The Carbocation (or carbocation-like) Electrophile
- Watch Out! Carbocation Rearrangements Can Occur In The Friedel-Crafts Alkylation Reaction
- If A Hydride Shift Or Alkyl Shift Will Result In A More Stable Carbocation, Assume It Will Happen
- Limitations of the Friedel-Crafts Alkylation
- Friedel-Crafts Acylation
- The Mechanism Of The Friedel-Crafts Acylation Reaction
- No Rearrangements Occur In The Friedel-Crafts Acylation
- Limitations of The Friedel-Crafts Acylation
- Summary: Friedel-Crafts Alkylation and Acylation
- Notes
- Quiz Yourself!
- (Advanced) References and Further Reading
1. Quick Recap On Electrophilic Aromatic Substitution: Onward To Carbon-Carbon Bond Forming Reactions!
This is the third in a series of three posts on the key electrophilic aromatic substitution (EAS) reactions in introductory organic chemistry.
- In Part 1 we covered halogenation (chlorination, bromination, and iodination) of aromatic rings via EAS.
- In Part 2 we covered nitration and sulfonylation of aromatic rings via EAS.
Taken together, so far we have learned reactions to form carbon-halogen, carbon-nitrogen, and carbon-sulfur bonds.
What important class of bond is missing so far?
Carbon-carbon bond forming reactions! [Note 1]
In this post, we’ll cover two important C–C bond-forming electrophilic aromatic substitution reactions which bear the names of their discoverers, Charles Friedel and James Crafts: Friedel-Crafts alkylation and Friedel-Crafts acylation.
We’ll also see that these reactions follow the familiar three-step pattern seen in previous electrophilic aromatic substitution reactions, namely:
- activation of electrophile with a Lewis acid
- attack of the “activated” electrophile by the aromatic ring
- deprotonation to restore aromaticity
2. Friedel-Crafts Alkylation Of Aromatic Rings
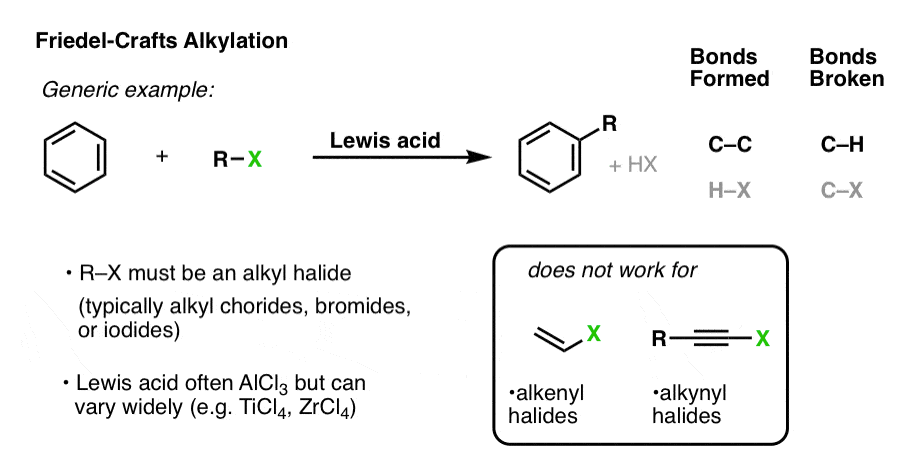
When an alkyl halide is treated with a Lewis acid in the presence of an aromatic ring, the alkyl group can be added to the ring (forming C-C) with the loss of a C-H bond. This electrophilic aromatic substitution reaction is known as the Friedel-Crafts alkylation reaction.
Generally, no reaction occurs in the absence of Lewis acid. A common choice for the Lewis acid is aluminum chloride, AlCl3 , but many others may be used, such as FeCl3 among others.
Alkyl halides (typically chlorides, bromides, and iodides) must be used, as the reaction fails completely for alkenyl and alkynyl halides.
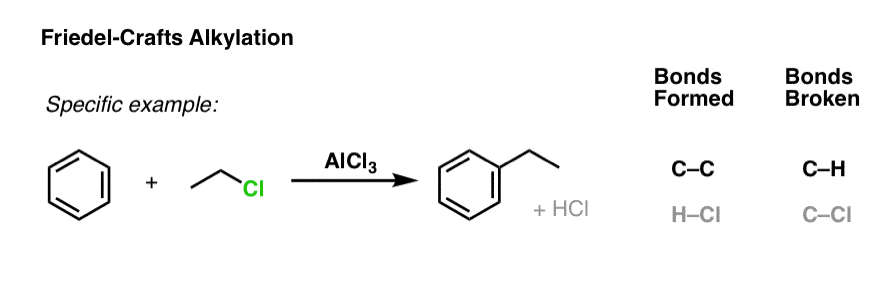
Here’s a specific example using ethyl chloride:
3. The Role of The Lewis Acid In Friedel-Crafts Alkylation Is To Activate The Alkyl Halide
If no reaction occurs in the absence of a Lewis acid, then what is the role of the Lewis acid here?
Like we saw in the two previous posts on electrophilic aromatic substitution reactions, Lewis acids “activate” the electrophile by coordinating to the leaving group, making it a weaker base, and a better leaving group (AlCl4– is a weaker base than Cl– ). The end result is that coordination of the Lewis acid to the electrophile makes the species a better electrophile.
For example, with isopropyl chloride (below), the first step is coordination of coordination of AlCl3 to the chlorine atom. This weakens the C-Cl bond, with the result that the Cl can depart (as AlCl4– ) to give a secondary carbocation (a better electrophile than isopropyl chloride itself).
- With secondary and tertiary halides, full dissociation to a carbocation can occur.
- In the case of primary (and methyl) alkyl halides, the electrophile is likely not a “free” carbocation, but a “carbocation-like” species where the C–Cl bond is considerably weakened/lengthened.
- As we mentioned briefly, no reaction occurs with alkenyl or alkynyl halides, largely because the carbocations of these species are so unstable and difficult to generate.
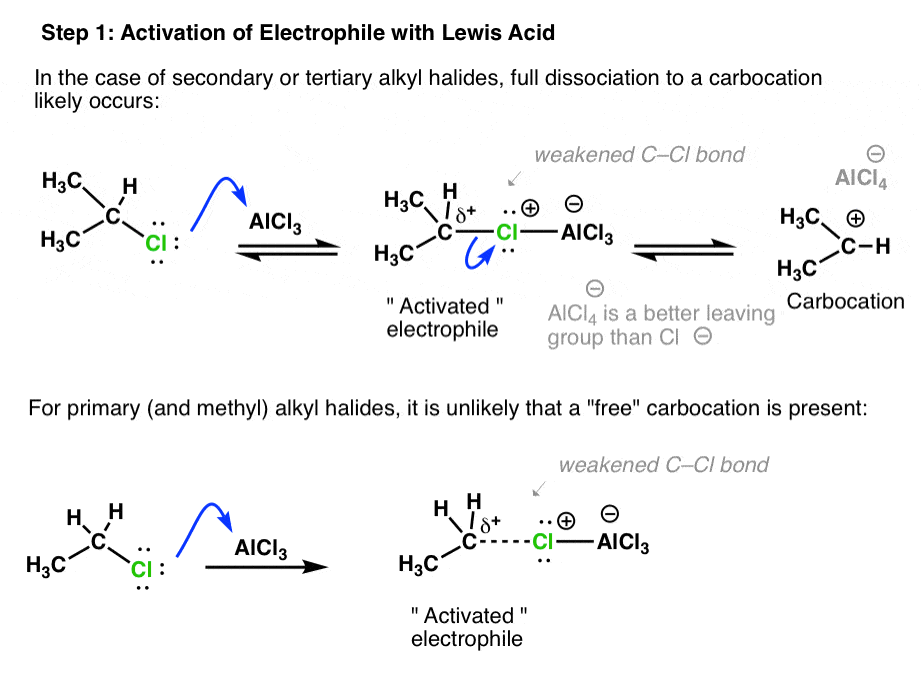
Note: that although here we are showing the carbocation electrophile in the Friedel-Crafts as being generated from an alkyl halide and a Lewis acid, there are other ways to generate a carbocation [such as through protonation of an alkene, see below]. We generally define the Friedel-Crafts alkylation as being the reaction of an aromatic ring with a carbocation (or carbocation-like) intermediate. See Note 2 for example.
4. The Key C-C Bond Forming Step Is Attack Of The Aromatic Ring Upon The Carbocation (or carbocation-like) Electrophile
Once the electrophile has been activated, the next step of the Friedel-Crafts is attack of the activated electrophile by the aromatic ring. This is also the rate-determining step, as it disrupts the aromaticity of the ring (and its ~36 kcal/mol of resonance energy).
In this step a C–C (pi) bond from the aromatic ring breaks, and a new C–C sigma bond is formed, leading to a carbocation intermediate:
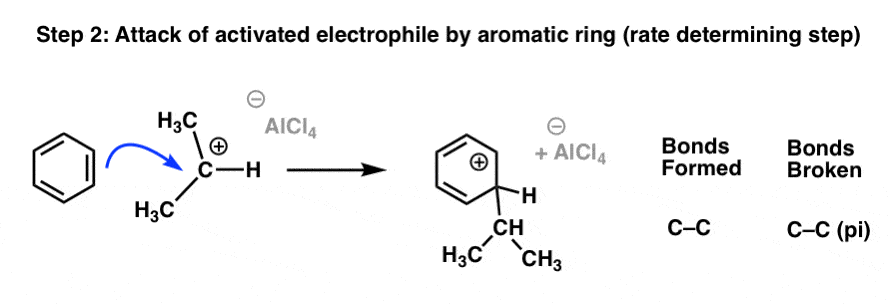
The last step is deprotonation of C–H by a weak base (e.g. Cl – ) to restore aromaticity at the ring:
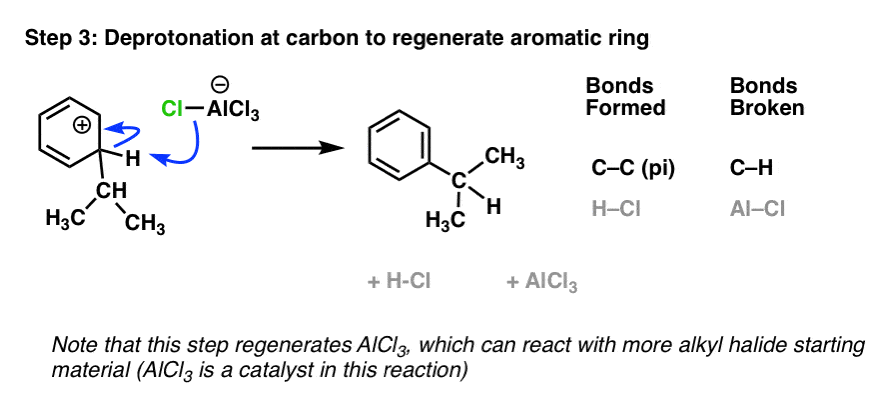
[another way to depict the curved arrows in this reaction is to dissociate Cl– from AlCl4– and then employ it as the base. Either way it works out to the same thing].
Note that AlCl3 is regenerated here, allowing it to be used again in step 1 with another equivalent of the alkyl halide. Hence, AlCl3 can act as a catalyst in this reaction, since it increases the rate of reaction but is not consumed by it.
5. Look Out! Carbocation Rearrangements Can Occur In The Friedel-Crafts Alkylation Reaction
Many university science courses are taught in units, where what you learn in one module has pretty much zero overlap with what you learn in another.
Needless to say, organic chemistry is not like this. You’ve probably already experienced a situation where concepts you learned in Org 1 reverberate back to Org 2 chapters in unexpected ways. Well, get ready for another fun example.
We showed above how ethyl chloride reacts with benzene and AlCl3 in the Friedel-Crafts alkylation to provide ethylbenzene.
Extension of this reaction from ethyl chloride to propyl chloride should correspondingly give propylbenzene.
Right?
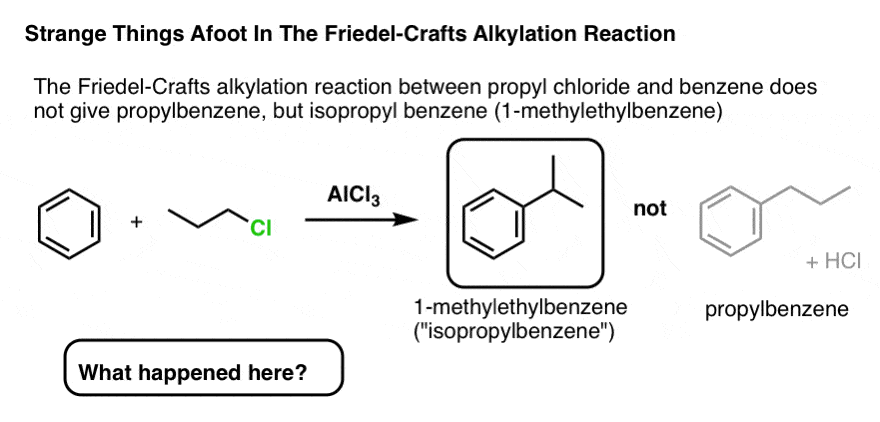
What the…. isopropylbenzene?
How did this happen?
Quick trip down memory lane. Remember this beloved reaction from Org 1?
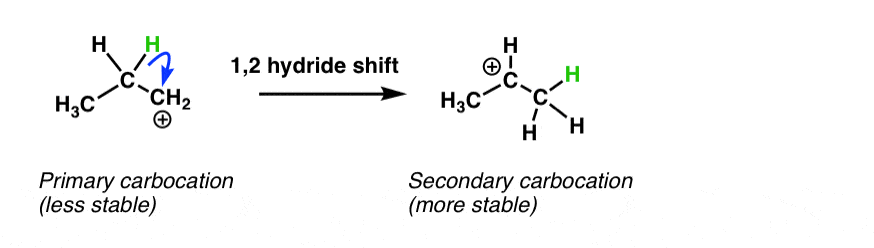
Ah, the hydride shift. Carbocations can rearrange via hydride and alkyl shifts such that a less stable carbocation is transformed into a more stable carbocation.
In the Friedel-Crafts, we’ve seen that coordination of a Lewis acid to an alkyl halide resulted in a carbocation (or in the case of primary alkyl halides, at least a “carbocation-like” species) that is then attacked by the aromatic ring in the rate-determining step.
6. If A Hydride Shift Or Alkyl Shift Will Result In A More Stable Carbocation, It Will Happen
So what is happening here is really no different: if a carbocation can rearrange to a more stable carbocation through a hydride or alkyl shift, it will do so.
Organic chemistry 2: the course where first-semester concepts come back to bite you in the ass.™
Here’s what happens in the case of propyl chloride.
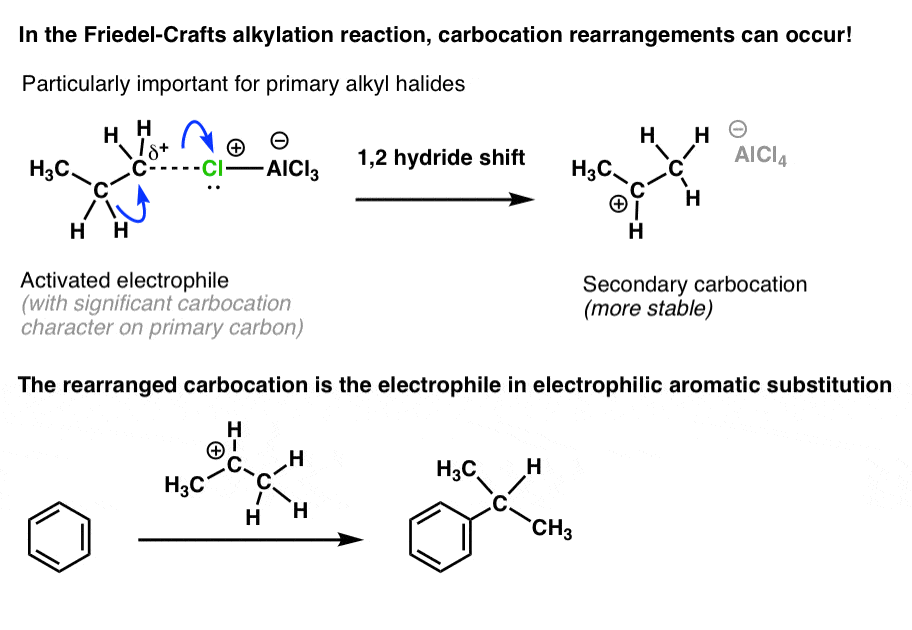
A shift of hydride from C2 to C1 results in a secondary carbocation, which is then attacked by the aromatic ring.
Bottom line for the Friedel-Crafts alkylation reaction:
- assume the alkyl halide goes through a carbocation
- assume that if the carbocation can rearrange to form a more stable carbocation through a hydride (or alkyl) shift, it will.
Another example of a rearrangement in the FC alkylation included in the footnotes just for fun. [Note 3]
7. Limitations of the Freidel-Crafts Alkylation
Final note on the Friedel-Crafts alkylation: a few drawbacks.
- First, as we’ve seen, carbocation rearrangements can occur. [There are ways of circumventing this issue indirectly, which we’ll hint at below [skip to bottom].
- Second, the Friedel-Crafts alkylation tends not to work well with electron-poor aromatic rings, particularly strongly deactivating substituents such as CF3, NO2, SO3H, and so forth. Halogens are OK.
- Third – and this is more of a practical issue than anything else, so is often ignored – the product of the FC alkylation is often a better nucleophile than the starting material. (Recall that alkyl groups are activating.) The result can be a bit like the Cookie Monster in a Chips Ahoy! factory – it can’t stop at just one, resulting in multiple alkylations.
8. Friedel-Crafts Acylation
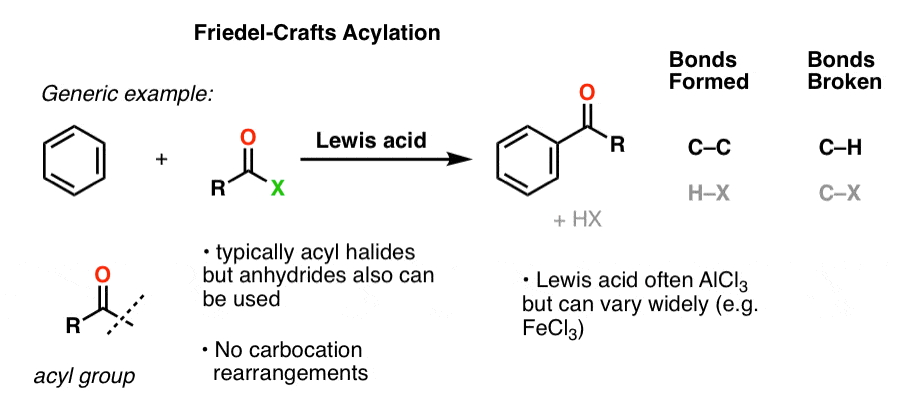
A process related to the Friedel-Crafts alkylation, called Friedel-Crafts acylation, was discovered by Friedel and Crafts around the same time (1877). If a Lewis acid is added to an acyl halide in the presence of an aromatic ring, an electrophilic aromatic substitution reaction can occur whereby the acyl group adds to the aromatic ring (with loss of HX).
As with the F.C. alkylation, the specific Lewis acid in the Friedel-Crafts acylation can vary. Aluminum chloride (AlCl3) is often used, but FeCl3 and other Lewis acids will also do the job.
Here’s a general example of the Friedel-Crafts acylation:
A specific example is the reaction between acetyl chloride and benzene catalyzed by aluminum chloride:
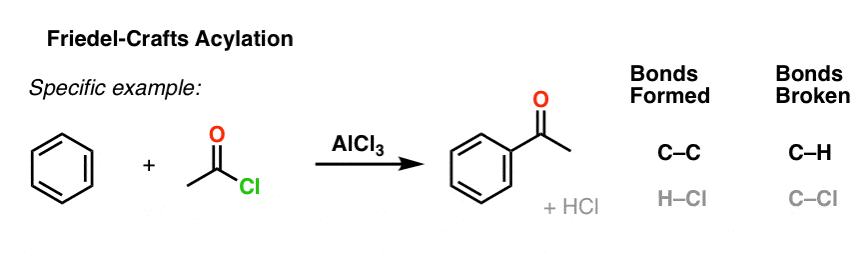
9. The Mechanism Of The Friedel-Crafts Acylation Reaction
So how does the Friedel-Crafts acylation reaction work?
As with FC alkylation, the first step is activation of the electrophile. Lewis acid coordinates to the halogen, and departure of the halogen (as AlCl4–) results in a fairly stable, resonance-stabilized carbocation know as the “acylium ion”.
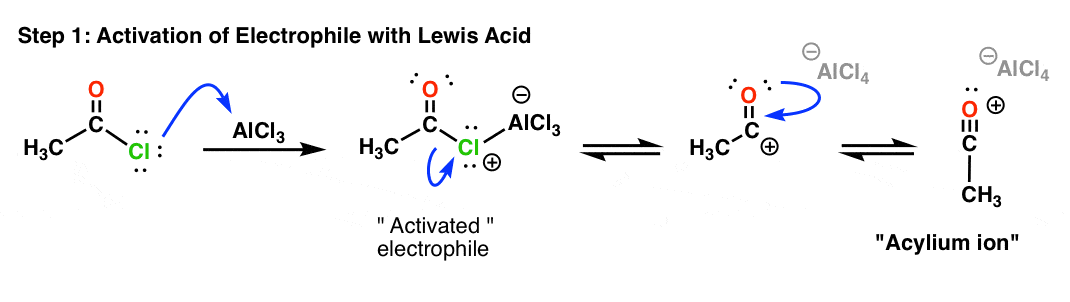
The acylium ion is the active electrophile in the Friedel-Crafts acylation reaction. Once formed, the acylium ion is attacked by the aromatic ring:
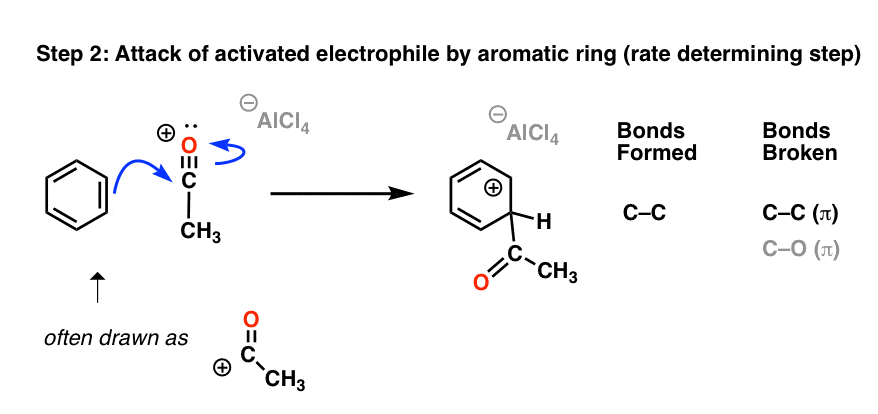
As with the Friedel-Crafts alkylation, the final step is deprotonation at carbon to regenerate the aromatic ring.
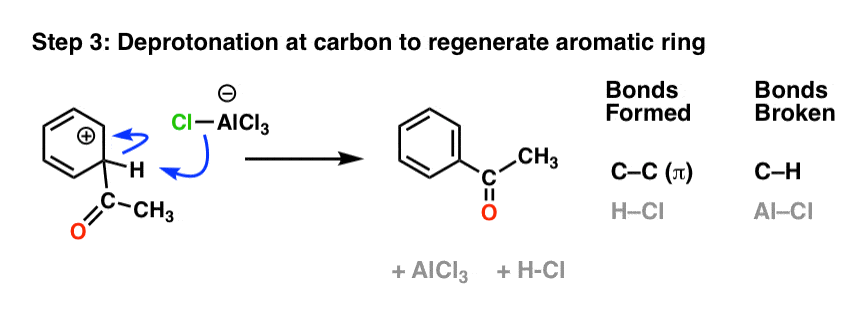
10. No Rearrangements Occur In The Friedel-Crafts Acylation
Unlike the Friedel-Crafts alkylation, no rearrangement occurs with the Friedel-Crafts acylation.
This opens up a “workaround” to use the Friedel-Crafts acylation to obtain products that are otherwise difficult to obtain through the Friedel-Crafts alkylation due to carbocation rearrangements. (We’ll talk about this in detail in a future article, but here we’ll just give a taste).
For instance, let’s look at how we could use this to produce propylbenzene, which we saw could not be made from the Friedel-Crafts alkylation reaction of benzene with AlCl3 and 1-propylchloride.
The first step here is to perform a Friedel-Crafts acylation reaction between benzene and propionylchloride, perhaps catalyzed by AlCl3. This gives us ethyl phenyl ketone.
The next step is to perform a reduction of the ketone to an alkane, which (as we’ll soon see) can be performed in various ways. This gives us 1-propylbenzene.
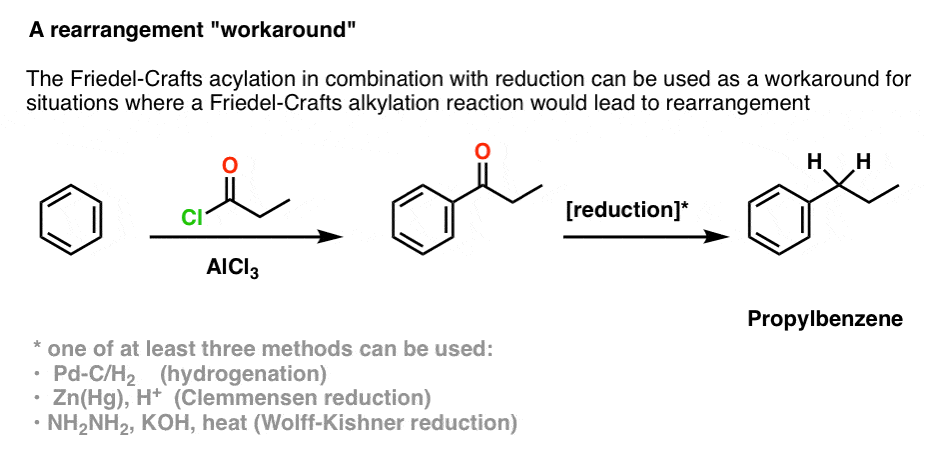
[Commenter Victor, from the Chemistry Help Center, helpfully notes that there is a fourth way of doing it – converting the ketone to a thioketal, and then reducing it down to the alkane with Raney nickel. ]
We will talk more about synthetic pathways for aromatic molecules in a future post.
11. Limitations of The Friedel-Crafts Acylation
- Similarly to alkylation, Friedel-Crafts acylation tends to fail on aromatic rings with strongly deactivating groups such as nitro, CF3, sulfonyl and so on. Halogenated aromatics still work, however.
- Put this in the “probably don’t need to know category”, but catalyst turnover in the Friedel-Crafts acylation isn’t great. In “real life”, a stoichiometric amount of AlCl3 is generally required since the AlCl3 coordinates strongly to the ketone product.
12. Summary: Friedel-Crafts Alkylation and Acylation
Since they form carbon-carbon bonds, the Friedel-Crafts alkylation and acylation reactions are particularly important electrophilic aromatic substitution reactions. Together with bromination, chlorination, nitration, and sulfonylation they round out the six core electrophilic aromatic substitution reactions.
Before we finish our treatment of electrophilic aromatic substitution, it’s worth going into detail on one more facet of the Friedel-Crafts that often gives students headaches; the intramolecular versions.
Next Post: Intramolecular Friedel-Crafts Reactions
Notes
Note 1. Bonus points if you said “carbon-oxygen” as a type of bond we haven’t seen formed in EAS. Direct electrophilic oxygenation of benzene rings is tricky to do in the lab. For our purposes, forming C-O on an aromatic ring is usually done indirectly, by means other than a direct EAS. Two ways we’ll explore in due course are the Baeyer-Villiger oxidation and certain reactions of diazonium salts.
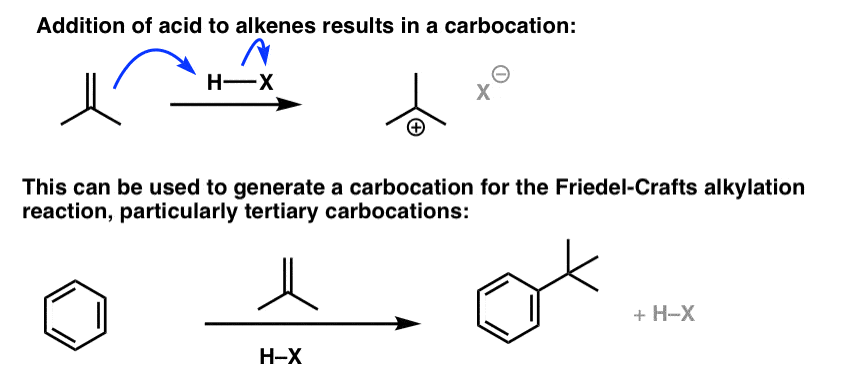
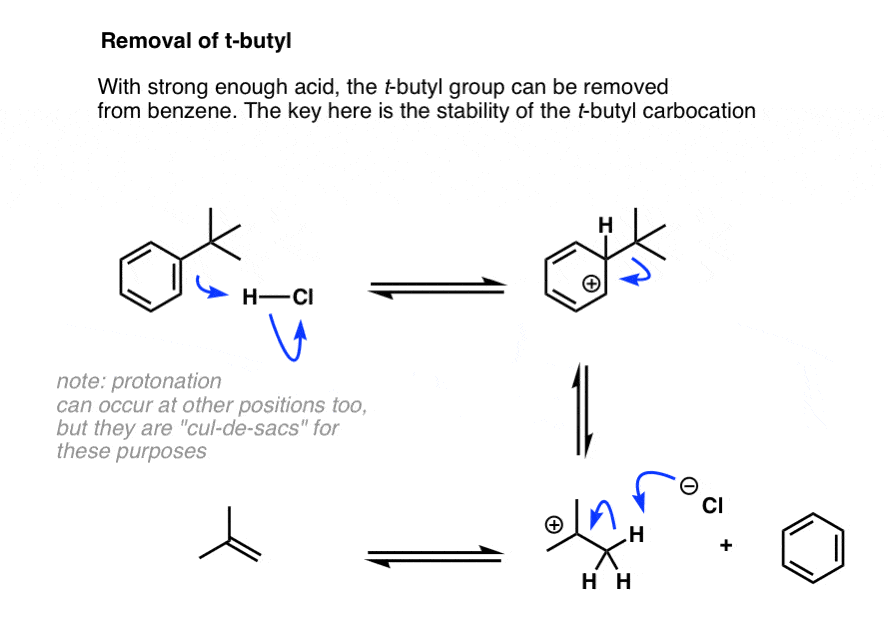
Note 2. Another way of performing a Friedel-Crafts alkylation is to generate the carbocation through protonation of an alkene. This works best when a fairly stable carbocation is generated, such as the t-butyl carbocation generated through protonation of 2-methylpropene.
Note 3. Bonus example with alkyl shift.
Note 4. Last post we learned that sulfonyl groups can be removed with strong acid, and I alluded to another group that can be removed that would be covered in the next post (i.e. this post). That group is t-butyl, which can be removed under forcing conditions, with strong acid. This works because the t-butyl carbocation is relatively stable and the reverse of the Friedel-Crafts alkylation is therefore feasible.
Quiz Yourself!
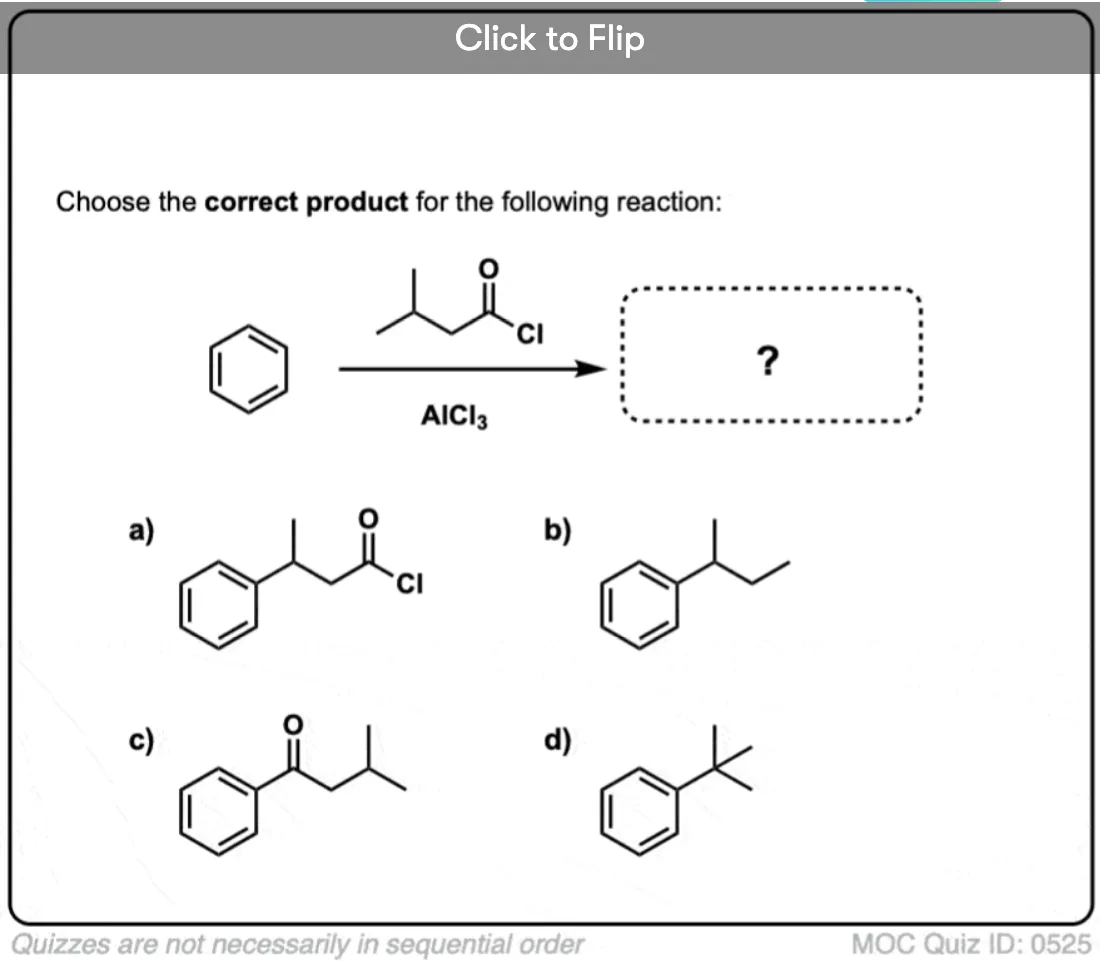
Become a MOC member to see the clickable quiz with answers on the back.
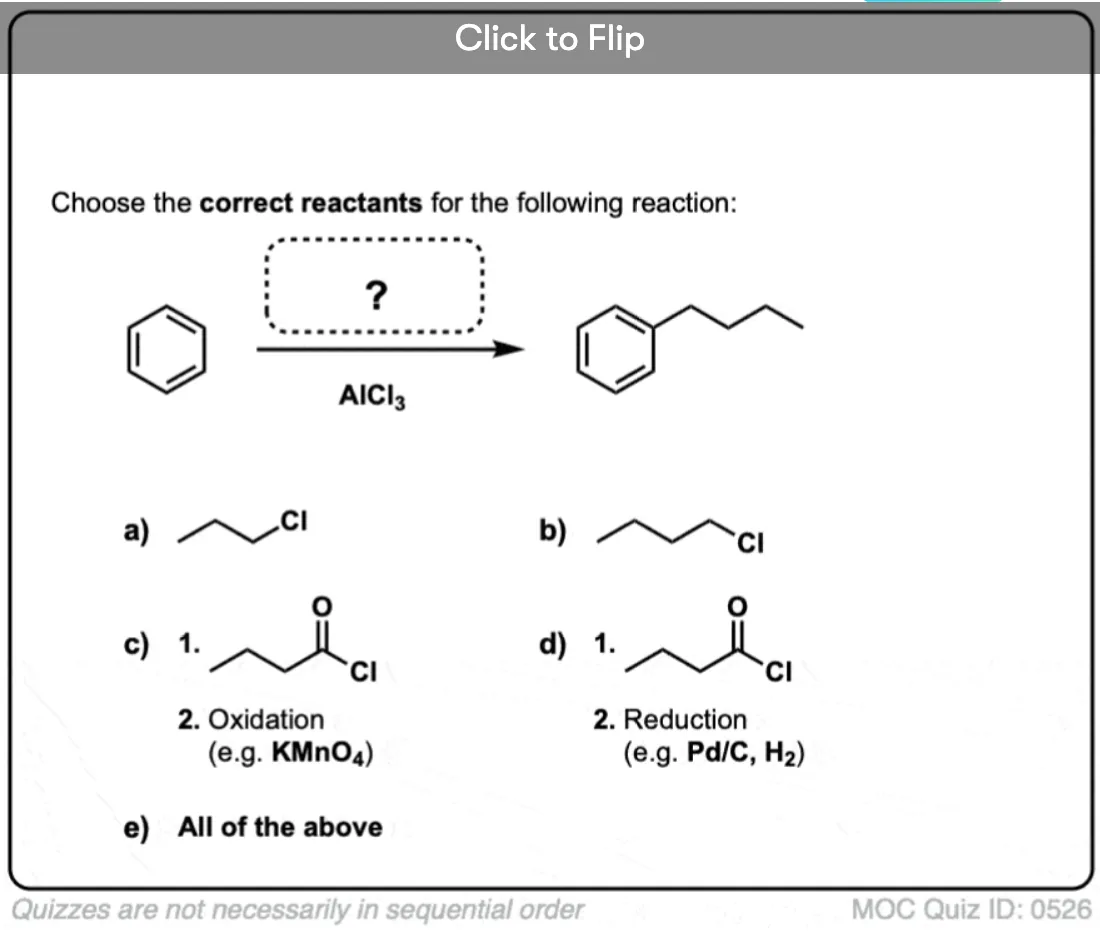
Become a MOC member to see the clickable quiz with answers on the back.
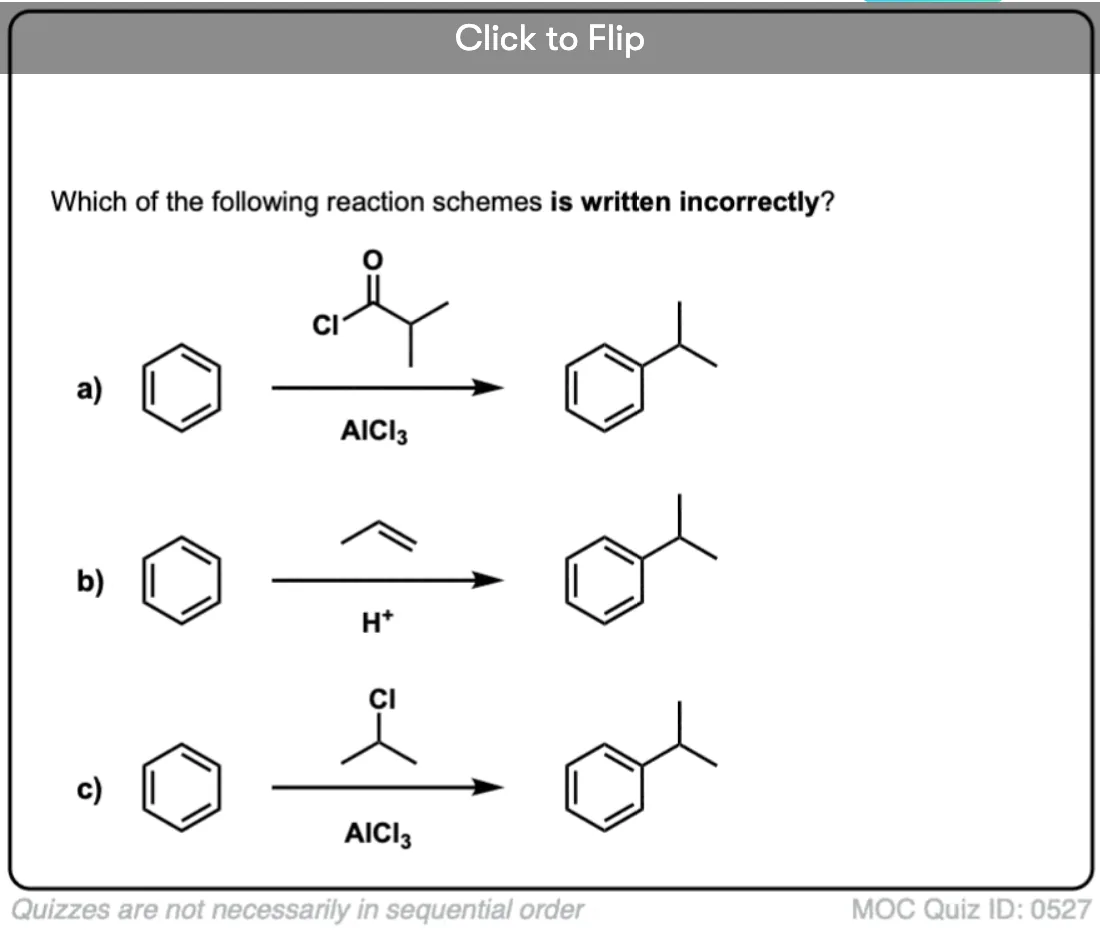
Become a MOC member to see the clickable quiz with answers on the back.

Become a MOC member to see the clickable quiz with answers on the back.
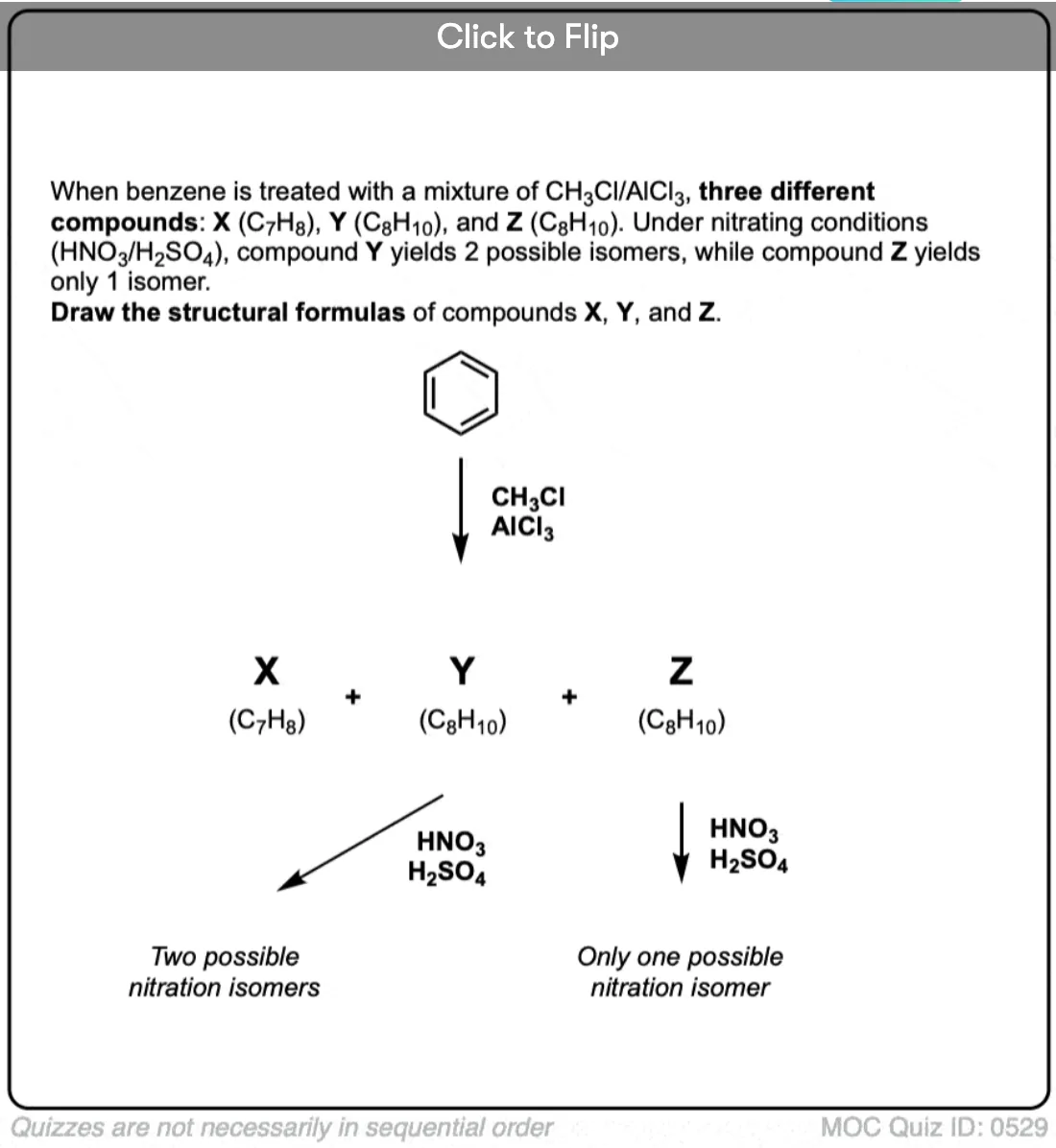
Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading
Friedel-Crafts alkylation:
- Jie Jack Li describes in his book Name Reactions:
“The discovery of the Friedel–Crafts reaction was the fruit of serendipity and keen observation. In 1877, both Friedel and Crafts were working in Charles A. Wurtz’s laboratory. In order to prepare amyl iodide, they treated amyl chloride with aluminum and iodide using benzene as the solvent. Instead of amyl iodide, they ended up with amylbenzene! Unlike others before them who may have simply discarded the reaction, they thoroughly investigated the Lewis acid-catalyzed alkylations and acylations and published more than 50 papers and patents on the Friedel–Crafts reaction, which has become one of the most useful organic reactions.”
DOI: 10.1007/978-3-642-01053-8_101 - Sur une nouvelle methode generale de synthese d’hydrocarbures, d’acetones, etc.
Friedel and J. M. Crafts
Compt. Rend. 1877 84, 1392-1395
The classic, original paper in French by Friedel and Crafts on the alkylation of aromatics (benzene in this case) with alkyl chlorides with AlCl3. - REARRANGEMENTS IN THE FRIEDEL-CRAFTS ALKYLATION OF BENZENE
HENRY GILMAN and R. N. MEALS
The Journal of Organic Chemistry 1943, 08 (2), 126-146
DOI: 10.1021/jo01190a003
This paper by the legendary American Chemist Prof. Henry Gilman (Iowa State) carefully studies the alkylation of benzene by long-chain alkyl halides (C10 and above). This is a monumental effort, especially considering this was prior to modern chromatographic or spectroscopic techniques (e.g. GC or NMR) that would make analysis of mixtures and characterization of these compounds so much easier.George A. Olah, who received the Nobel Prize in Chemistry in 1994, was well-known for his work in superacid and Friedel-Crafts chemistry. Here are a selection of his papers relevant to Friedel-Crafts alkylation: - Aromatic Substitution. VI. Intermediate Complexes and the Reaction Mechanism of Friedel-Crafts Alkylations and Acylations
G. A. Olah and S. J. Kuhn
Journal of the American Chemical Society 1958, 80 (24), 6541-6545
DOI: 10.1021/ja01557a022 - Aromatic Substitution. XVI.1 Friedel-Crafts Isopropylation of Benzene and Methylbenzenes with Isopropyl Bromide and Propylene
George A. Olah, Sylvia H. Flood, Stephen J. Kuhn, Maryanne E. Moffatt, and Nina A. Overchuck
Journal of the American Chemical Society 1964, 86 (6), 1046-1054
DOI: 1021/ja01060a016 - Aromatic Substitution. XVIII.1 Friedel-Crafts t-Butylation of Benzene and Methylbenzenes with t-Butyl Bromide and Isobutylene
George A. Olah, Sylvia H. Flood, and Maryanne E. Moffatt
Journal of the American Chemical Society 1964, 86 (6), 1060-1064
DOI: 1021/ja01060a018 - Aromatic Substitution. XIX.1 Friedel-Crafts Isopropylation and t-Butylation of Halobenzenes
George A. Olah, Sylvia H. Flood, and Maryanne E. Moffatt
Journal of the American Chemical Society 1964, 86 (6), 1065-1066
DOI: 1021/ja01060a019 - Aromatic Substitution. XXV.1 Selectivity in the Friedel-Crafts Benzylation, Isopropylation, and t-Butylation of Benzene and Toluene
George A. Olah and Nina A. Overchuk
Journal of the American Chemical Society 1965, 87 (24), 5786-5788
DOI: 1021/ja00952a047
The first sentence in this paper is of note: “Friedel-Crafts alkylations are notorious for their unreliable kinetic behavior”. This is because they are largely heterogeneous, or occur in 2 phases. Also, the footnote indicates that inquiries should be addressed to the Department of Chemistry at the Western Reserve University, Cleveland, OH – before it merged and became Case Western Reserve University. - Aromatic substitution. XXVIII. Mechanism of electrophilic aromatic substitutions
George A. Olah
Acc. Chem. Res., 1971, 4 (7), 240-248
DOI: 10.1021/ar50043a002
An account by Prof. Olah on the work he had carried out studying the mechanism of various types of electrophilic aromatic substitution reactions – nitration, halogenation, as well as Friedel-Crafts acylation and alkylation. - Aromatic substitution. XXXVII. Stannic and aluminum chloride catalyzed Friedel-Crafts alkylation of naphthalene with alkyl halides. Differentiation of kinetically and thermodynamically controlled product compositions, and the isomerization of alkylnaphthalenes
George A. Olah and Judith A. Olah
Journal of the American Chemical Society 1976, 98 (7), 1839-1842
DOI: 1021/ja00423a032
This is a similar paper by Prof. Olah and his wife, Judith Olah, on the mechanism of Friedel-Crafts alkylation, except using naphthalene instead of benzene. Naphthalene is different in that there are two sites for monosubstitution – the a and b positions. - Friedel-Crafts alkylation of anisole and its comparison with toluene. Predominant ortho-para substitution under kinetic conditions and the effect of thermodynamic isomerizations
George A. Olah, Judith A. Olah, and Toshiyuki Ohyama
Journal of the American Chemical Society 1984, 106 (18), 5284-5290
DOI: 1021/ja00330a042
A surprising sentence in this paper: “No systematic study of the alkylation of anisole was, however, yet reported. Consequently we undertook such a study and report our results”. Sometimes science has these low-hanging fruit, and thoroughly reading the literature can lead you to them.The following papers are related to dealkylation, isomerization, or transfer alkylation under Friedel-Crafts conditions:Friedel-Crafts acylation: - DESOXYBENZOIN
F. H. Allen and W. E. Barker
Org. Synth. 1932, 12, 16
DOI: 10.15227/orgsyn.012.0016
A fairly representative experimental procedure for a Friedel-Crafts acylation in Organic Syntheses, a well-known source for reliable, independently tested synthetic organic experimental laboratory procedures. The product desoxybenzoin would be better known today as dihydrochalcone. - Aromatic Substitution. XXII. Acetylation of Benzene, Alkylbenzenes, and Halobenzenes with Methyloxocarbonium (Acetylium) Hexafluoro- and Hexachloroantimonate
George A. Olah, Stephen J. Kuhn, Sylvia H. Flood, and Barbara A. Hardie
Journal of the American Chemical Society 1964, 86 (11), 2203-2209
DOI: 1021/ja01065a020
This paper deals with the acetylation of aromatics with preformed CH3CO+ salts, which Prof. Olah figured how to isolate using SbF5. Also, note where these papers were submitted from – Prof. Olah fled from Hungary in the 1950’s to Canada and joined the Dow Chemical Company there. - Aromatic substitution. XXIX. Friedel-Crafts acylation of benzene and toluene with substituted acyl halides. Effect of substituents and positional selectivity
George A. Olah and Shiro Kobayashi
Journal of the American Chemical Society 1971, 93 (25), 6964-6967
DOI: 1021/ja00754a045
This is a mechanistic study of Friedel-Crafts acylation (or ‘benzoylation’ in this case) using the Hammett approach, a classic tool in physical organic chemistry. More reactive electrophiles have lower kt/kb ratios and low o/p selectivity, while less reactive electrophiles have higher kt/kb ratios and high o/p selectivity. kt/kb in this case refers to the relative rate of reacting with toluene vs. benzene.Oxygenation (or oxyfunctionalization) of hydrocarbons and aromatics is definitely possible with the right conditions, as the following two papers describe. Prof. George Olah has written a series of papers on the subject. - Oxyfunctionalization of hydrocarbons. 8. Electrophilic hydroxylation of benzene, alkylbenzenes, and halobenzenes with hydrogen peroxide in superacids
George A. Olah and Ryuichiro Ohnishi
The Journal of Organic Chemistry 1978, 43 (5), 865-867
DOI: 1021/jo00399a014 - Oxyfunctionalization of hydrocarbons. 14. Electrophilic hydroxylation of aromatics with bis(trimethylsilyl) peroxide/triflic acid
George A. Olah and Thomas D. Ernst
The Journal of Organic Chemistry 1989, 54 (5), 1204-1206
DOI: 1021/jo00266a041
In this paper, bis(trimethylsilyl)peroxide (TMSOOTMS) is used as the oxidant.
‘,’EAS Reactions (3) – Friedel-Crafts Acylation and Friedel-Crafts Alkylation
Yes, that is true
We’ve read that aniline and phenols dont give friedel crafts as well because they form a complex with alcl3 on the nitrogen or oxygen due to the presence of a lone pair.
Acetophenone+Alcl3+Aq.HCl -A+B
Can you name the reason and give the products
do methyl ketone reacts with benzene in the presence of AlCl3
No, not through any kind of Friedel-Crafts reaction.
Thanks for replying to my question!
I have a few doubts about the mechanism that you have drawn out for the double Friedel-Crafts reaction. In the mechanism, the AlCl3 attacks the carbonyl oxygen atom instead of the oxygen atom which is part of the ring, which is different from how AlCl3 doesn’t attack the carbonyl oxygen atom when we are using acyl halides. Is there a reason why the mechanism is different in this case? And also, if we use the same epoxide ring as before, but without the carbonyl oxygen atoms, will the Friedel-Crafts reactions still take place?
Also, as AlCl3 is a catalyst, we should be getting back AlCl3 by the end of the reactions. However, in your mechanism, we are getting [OAlCl3]− instead. So, shouldn’t the oxygen atom still remain on the carbon chain, resulting in the formation of a carboxylic acid just before the cyclization of the carbon chain occurs?
Hi! I have 2 questions:
1)As you have pointed out, carbocation rearrangements can occur in Friedel-Crafts Alkylation reactions. However, this seems to only occur when AlCl3 is used as a catalyst. When FeCl3 is used in your given example, isopropylbenzene becomes a minor product and propylbenzene becomes the major product. Is there a reason why this occurs?
Related links: https://tinyurl.com/4jy6kfea , https://tinyurl.com/yjm9smc9
2)In this question (https://tinyurl.com/bddnkd97) in the reaction from P to Q, I am not sure how AlCl3 attaches to the Epoxide and how the resulting Friedel-Crafts reaction takes place and I was wondering whether you could explain how this mechanism takes place?
1) I don’t think there is anything magical about FeCl3. Friedel Crafts rearrangements will happen as soon as heat is applied.
2) Hi, yes, I have drawn out the mechanism for this double Friedel-Crafts acylation reaction.https://cdn.masterorganicchemistry.com/wp-content/uploads/2022/08/Supp-1-Mechanism-for-intramolecular-Friedel-Crafts.gif
Organic chemistry 2: the course where first-semester concepts come back to bite you in the ass.™ Mr. James please don’t put such sentences.
Occasionally I can be irreverent.
Thank you for your concern to my query…Please tell me what is the role of Anhy AlCl3 in FC reaction of Benzoic acid?Does AlCl3 binds with COOH group…If so in what way…Happy to hear from you…
Please let me know the clear mechanism that why benzoic acid doesn’t undergo Friedel Crafts alkylation and acylation reaction… Thank you….
Friedel Crafts reactions generally fail when electron withdrawing groups are present on the ring. The carboxylic acid is an electron withdrawing group, and this renders the aromatic ring less electron rich (and therefore less nucleophilic).
I’m very much satisfied learning your organic chemistry concepts…really the master organic chemistry masters me in organic chemistry topics conveniently…
I request you that as I’m a teacher shall I use your content for my inline class room teaching… looking forward for your suggestion…Have a nice day 💐
I am a jee aspirant and ur content and article are beautifully explained……. thank u
In your acylation and alkylation mechanism, you list AlCl3 as one of the products, which implies that the reaction is catalytic. If that is the case, why do literature FC reactions always use stoichiometric or excess amounts of aluminum chloride?
Quote: “Put this in the “probably don’t need to know category”, but catalyst turnover in the Friedel-Crafts acylation isn’t great. In “real life”, a stoichiometric amount of AlCl3 is generally required since the AlCl3 coordinates strongly to the ketone product.”
I didn’t include the coordination of AlCl3 to the ketone in the schemes because I wanted to keep them as straightforward and uncluttered as possible.
What’s the product if for C6H5 + FCH2Cl ( BCl3 as Lewis acid) -> ?
What’s a better leaving group, Cl or F ?
Please include examples of other types of benzene molecules being used, especially substituted ones.
Thanks for the suggestion. For this post I wanted to just use simple examples. I suggest going to some of the other articles on synthesis for some slightly more complicated examples. The Reaction Guide also has more complex examples too.
Hi. How do you know which groups will deactivate a benzene ring to the point that a Friedel-Crafts reaction will not work? Is it ALL deactivating groups except halogens? I’ve struggled to find an overarching rule for this. Are these groups different for a alkylation versus an acylation F-C? What groups other than NH2, NHR, and NR2 are strong enough to prevent a Friedel-Crafts reaction?
There’s two factors going on. First, groups more electron withdrawing than the halides will make the ring so deactivated that EAS won’t happen. This has just been learned from vast trial-and-error, to be honest. Second, if groups are present that can act as nucleophiles (amines being the most prominent) they will react with the Friedel-Crafts reaction rather than the aromatic ring! For this reason, if your aromatic ring contains an amine, protect it as an amide and remove it later.
I would like to know the Friedel Crafts acylation works for CF3COCl or not.
Aromatic ring is activated with an electron donating group.
Couple of notes:
Minor: missed a negative charge on AlCl4 in step 3 of acylation when it acts as a base.
Major: When you discuss the reduction methods for the product of acylation, you list Kishner-Wolff, Clemmensen, *and* hydrogen on palladium??? as the ways to remove the C=O. The last one will only reduce it to an alcohol. If you wanted to use H2/Ni (not Pd still), you’d need to convert it into a thioacetal first.
Thank you for the typo correction! The hydrogenation works for benzylic ketones (and will reduce benzyl alcohols as well), but I wouldn’t do it on aliphatic ketones.
In contrast, the Raney Nickel reduction of the thioketal that you helpfully mentioned should work on pretty much any ketone.
As usual, great post. Some minor corrections needed, though:
– “The end result is that coordination of the Lewis acid to the electrophile makes the species is a better electrophile.” Probably should read “… makes the species a better electrophile.”
– “The first step here is to perform a Friedel-Crafts acylation reaction between benzene and 1-propylchloride, …” Should read: “… reaction between benzene and propionyl chloride, …”
– plus some other minor things, like double or unnecessary spaces, etc.
Thank you very much. Fixed the typos. I am in your debt!