Dienes and MO Theory
Bonding And Antibonding Pi Orbitals
Last updated: June 2nd, 2026 |
Bonding And Antibonding Orbitals For A Simple Pi Bond
- Two adjacent p-orbitals each containing an electron can overlap to form a pi-bond
- In a pi bond, the two atomic orbitals (p-orbitals) overlap to form two molecular orbitals (pi-orbitals)
- Overlap between two p-orbitals with the same phase results in constructive orbital overlap and allows the electrons to be shared between the two atoms, resulting in bonding and a lowering of energy
- Overlap between two p-orbitals with the opposite phase results in destructive orbital overlap and prevents the electrons from being shared between the two atoms. Repulsion between the two positively-charged atomic nuclei being held closely together results in a more energetically unstable situation called antibonding (π* )
- In the case of a simple pi-bond with two electrons, the bonding pi-orbital (π) is the highest-occupied molecular orbital (HOMO)and the antibonding orbital is the lowest-unoccupied molecular orbital (LUMO)
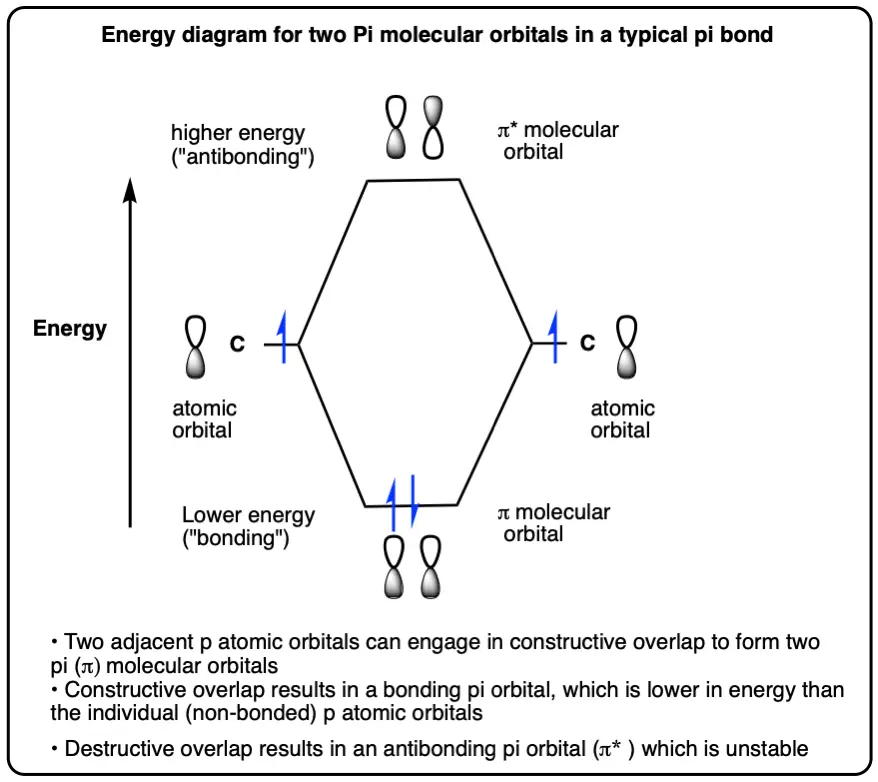
Table of Contents
- Relationship Energy Diagrams: “Bonding” and “Antibonding”
- The Full Relationship Energy Diagram
- Non-Bonding, Bonding And Antibonding Orbitals
- Energy Diagram Including Bonding, Non-Bonding, And Antibonding Orbitals
- “Why Do Antibonding Orbitals Even Exist” ?
- Molecular Orbitals Are Formed By The Overlap Of Atomic Orbitals
- Bonding Pi Molecular Orbitals Form Through The Side-On Constructive Overlap Of p Orbitals
- Antibonding Pi Molecular Orbitals Result From the Destructive Side-On Overlap Of Adjacent p Orbitals
- An Energy Diagram For A Simple Pi Bond Incorporating Bonding and Antibonding Pi Molecular Orbitals
- Conclusion: Some Rules for Pi Molecular Orbitals
- Notes
- Quiz Yourself!
1. Relationship Energy Diagrams: “Bonding” and “Antibonding”
In the previous post in this series, “On Conjugation and Resonance“, I warned that in order to gain a deeper understanding of some of the concepts ahead, we’re going to need to go into MO theory.
But before we do… shall we talk about dating? It’s Valentines day as I write this, after all…
A lot of people say they’re happy being single, and I believe that many likely are. But in the back of their mind of many single people is the thought that if they just found the right person, they might be even happier – or less unhappy, which is a crappy way to look at it psychologically but necessary if you wish to draw a diagram where a “happy couple” is occupying a “potential energy well”, below.
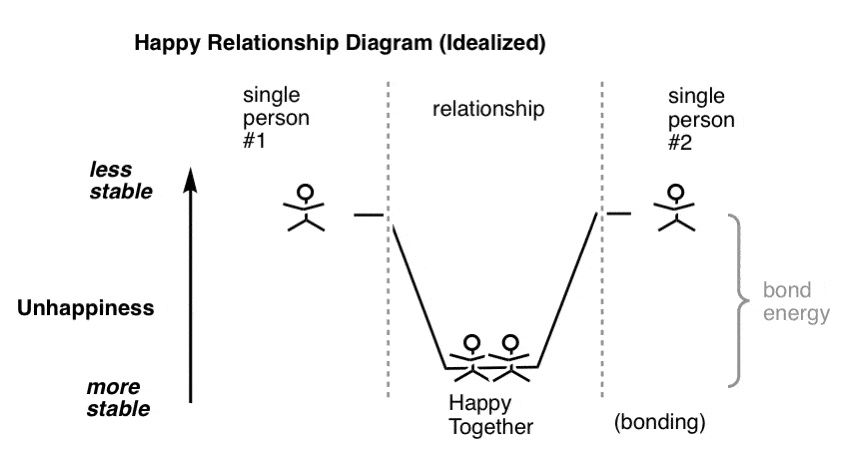
(can also be yenta-catalyzed)
The decrease in unhappiness brought about by bonding can be quantified as the “bond energy”.
Of course, it can take a lot of random collisions before two single people fall into the potential energy well that categorizes a happy relationship.
Worse, two people can suddenly find themselves in a relationship where they were initially attracted to each other, but upon being in close proximity realize that they were actually happier being single after all.
That gives you a situation like the one below which you can think of as “antibonding” – an unhappy, unstable couple, destined to separate back into its individual components.
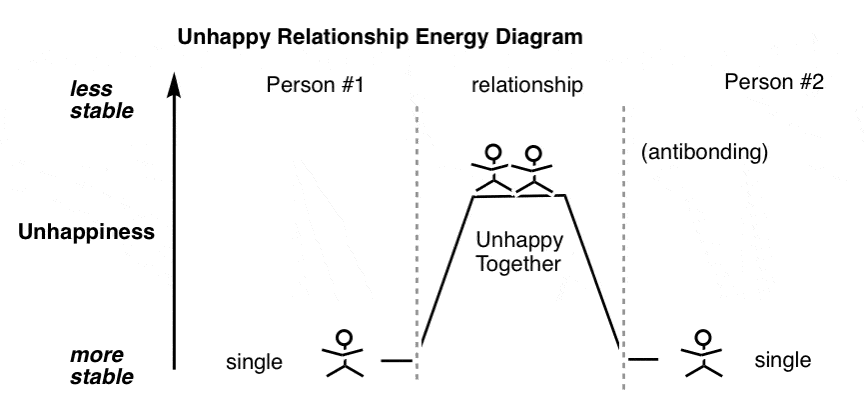
2. The Full Relationship Energy Diagram
The romantic view of love is that if the potential energy well is deep enough, you have Happily Ever After, lovers destined never to part.
Of course, that’s partly a reflection of the fact that romantic comedies cut out right after the wedding scene. They don’t check in six months later when Prince Charming has turned into “Mr. Never Takes His Kleenex Out of His Pants Before Putting Them In The Dryer” and Snow White has morphed into “Mrs. Keeps Leaving Her Disgusting Jammy Knife On The Kitchen Counter”.
The truth is that even happy relationships possess latent unhappiness. Strain them with enough force, and instability (and breakup) may result.
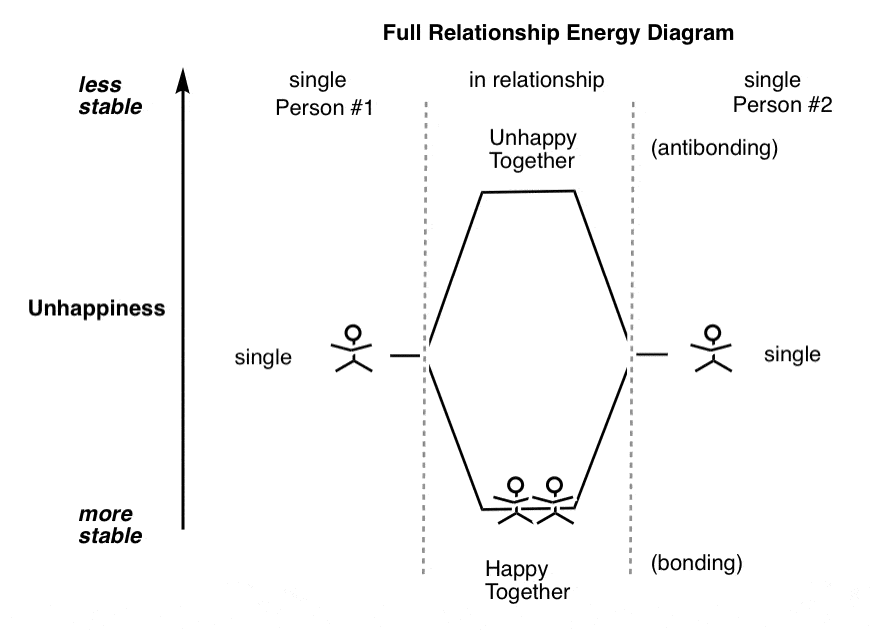
And this diagram is just for two people!
We don’t need to make drawings of the destabilization that can occur when a third person enters the relationship.
3. Non-Bonding, Bonding And Antibonding Orbitals
At the risk of being pedantic, let’s remind ourselves: why do bonds form in the first place? What is the reason why atoms would bother sharing their electrons with each other at all? Why can’t they just “be happy being single?”.
Some are, of course – the noble gases come to mind. But for elements in the first row of the periodic table with less than 8 valence electrons, particularly carbon, nitrogen, oxygen, and fluorine, there is a strong electrostatic driving force to acquire a full octet of valence electrons.
It may help to recall the forces at work. In a chemical bond, two negatively charged electrons are held between the two positively charged nuclei. If you use the Coulomb equation to model this, the attractive forces between the two electrons and the two nuclei (opposite charges attract, remember) outweigh the repulsive forces between the two positively charged nuclei and two negatively charged electrons, respectively. There is a net lowering of energy – a stabilization – imparted by bringing the nuclei together in this way. This lowering of energy is called the bond dissociation energy.
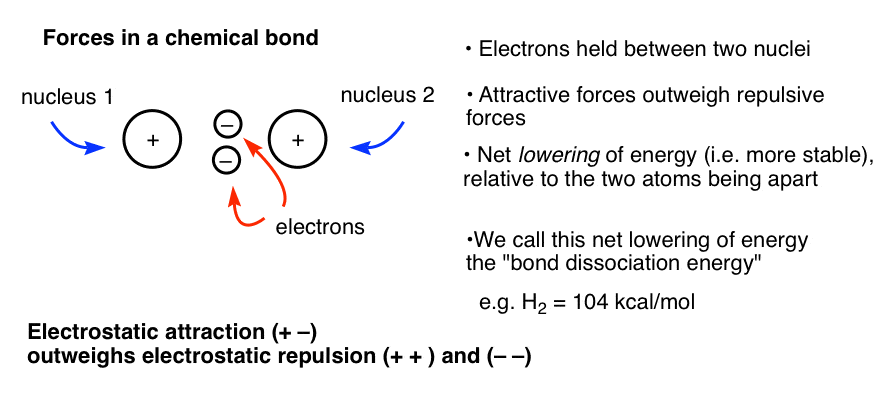
Now imagine another situation, where the nuclei are the same distance apart, but the electrons cannot be held between them (due to a node, see below). In this case, you have two positively charged nuclei held closely together in space but no electrons between them providing a stabilizing attractive interaction. The repulsion between the two nuclei (and the electrons with each other) outweighs any attractive forces between the electrons and nuclei, and the result is more unstable than the situation for two non-bonded atoms. We call this situation “antibonding”.
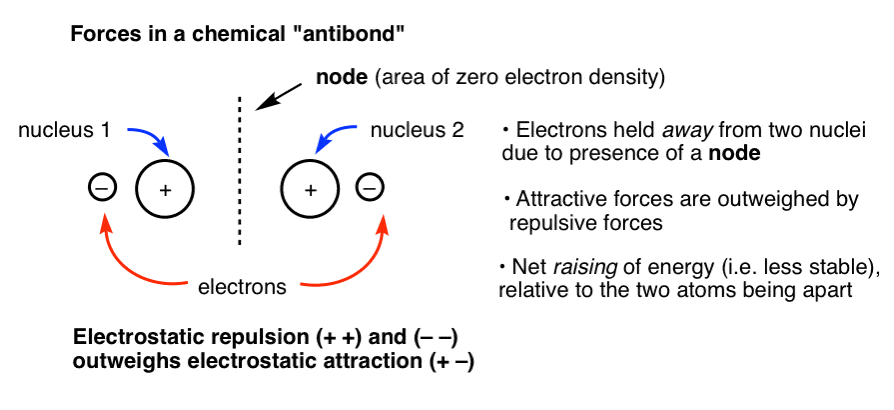
4. Energy Diagram Including Bonding, Non-Bonding, And Antibonding Orbitals
So, like relationships, in chemical bonding we’ve seen three situations of differing energy.
- “nonbonding” – the energy of the atom in the absence of any bonding.
- “bonding” – the energy in the happy situation where the two electrons are between the atoms, which is more stable: attraction > repulsion
- “anti bonding” – the energy in the unhappy situation where the electrons are held away from the atoms: repulsion > attraction
For bonding between two identical atoms, we can roughly sketch out these three levels as follows. [Note 2]
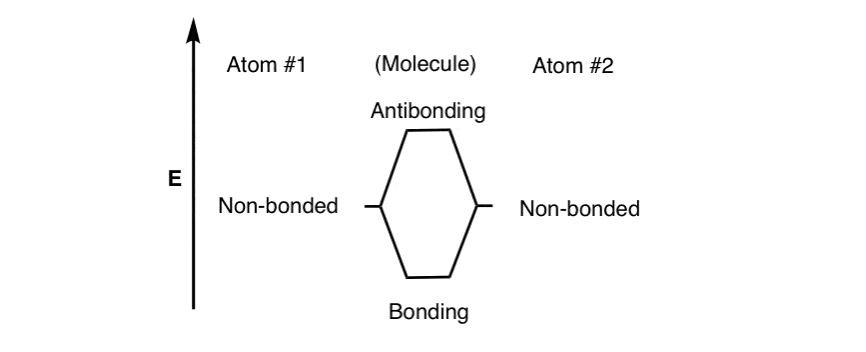
Note that we haven’t populated the levels with electrons yet. That comes next.
5. “Why Do Antibonding Orbitals Even Exist” ?
“Why does anti bonding even exist?”, you might wonder. Why do we have to concern ourselves with a situation that is even more unstable than for two atoms being apart?
As we’ll see, it’s quite common for electrons to occupy antibonding states – albeit for brief periods of time – without the molecule being destroyed. Electrons are small (1/1840 the mass of a proton) and nuclei are heavy; an electron can zip up to an antibonding state and back down to the bonding orbital in much less time than it would take for the bond to dissociate. [Note 1]
In fact, every time you observe the green leaves of a plant or the petals of a flower, what you are seeing is the result of visible light (of frequency ν) being absorbed by a chromophore (e.g. chlorophyll), promoting an electron from a pi (bonding) orbital to a pi* (antibonding) orbital, with a gap in energy E = hν. The high-energy electron can then just relax back to ground state, emitting a photon in the process, and the molecule is back to where it started.
As for “why antibonding exists”, that’s an excellent question.
No electrons may have the exact same quantum number. A bonding orbital can hold two electrons, each with opposite spin, and that’s it. Polygamy may be a thing in some cultures, and “alternative lifestyles” abound, but Wolfgang Pauli’s exclusion principle is more unbreakable than any religious or cultural ordinance.
By “quantum numbers” , we mean (n, l, m, and spin), the parameters of the Schrödinger wave equation which describes the energy and positions of electrons. What we call “orbitals” are actually 3-dimensional shapes where there is a 95% probability of an electron being found. Each orbital (a unique combination of n, l, and m) can accommodate a pair of electrons with opposite spin, +1/2 and –1/2.
Hence, when we populate our energy diagram with electrons, the most that any individual orbital can accommodate is two.
The Aufbau principle states that the lowest energy orbitals are filled first. It’s a bit like a Greyhound bus; people only stand in the aisles once the seats fill up.
So with two electrons (represented by half-arrows) to populate the orbitals, our sketch would look like this. Note the opposite spins in the bonding (molecular) orbital. For our singly-occupied orbitals, the orientation of the electron spins are generally drawn as being the same (a nod to Hund’s rule)
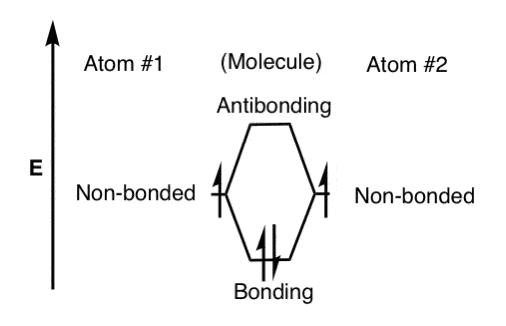
Were a third electron to be added to this, we’d have to put it in an anti bonding orbital (unstable). And were a fourth electron to be added, we’d have a doubly populated anti bonding orbital, which is extremely unstable (and a pretty good description of why helium doesn’t form He2 ).
6. Molecular Orbitals Are Formed By The Overlap Of Atomic Orbitals
The orbitals we generally concern ourselves with in introductory organic chemistry are the s and p orbitals.
s orbitals generally have the appearance of spheres. p orbitals look like dumbbells. d and f orbitals have a variety of fascinating shapes that we won’t talk about here.
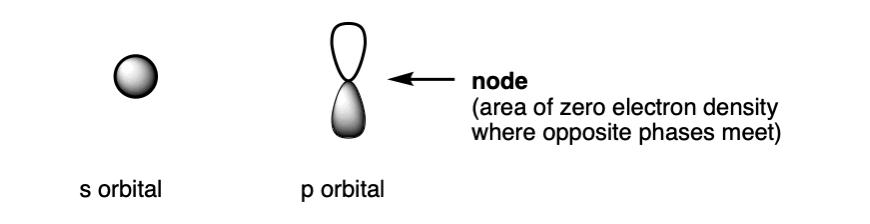
That dot at the centre of the p orbital is what’s called a node, an area of zero electron density, where there is a transition between phases. For the 2p orbital, the node resides directly at the nucleus. Fun fact: the number of nodes increases with the principal quantum number, n. The 2s and 2p orbitals each have one node, the 3s and 3p two, and so on.
When atomic orbitals overlap, they form molecular orbitals: areas of space where electrons are shared between atoms.
When s-0rbitals overlap with each other (or hybrid orbitals with s-character, such as sp3, sp2, and sp), sigma ( σ) molecular orbitals are formed. This is “end-on” overlap. The bonding in hydrogen (H2) is a perfect example.
The number of orbitals is never changed by bonding. The number of molecular orbitals always equals the number of contributing atomic orbitals. Hence for H2, built from two atomic (1s) orbitals, we obtain two molecular orbitals (bonding and anti bonding). Bonding in CH4, built from 4 hydrogen 1s orbitals and 4 hybrid carbon sp3 orbitals, has 8 molecular orbitals in total.
7. Bonding Pi Molecular Orbitals Form Through The Side-On Constructive Overlap Of p Orbitals
In this series of posts, we’ve been discussing Pi (π)bonding, which is the interaction (overlap) between two p orbitals. Bonding in this case is not end-on as in sigma bonding, but “side-on”, which as we saw last time, is responsible for the rotation barrier in alkenes.
When two p (atomic) orbitals overlap, we form two Pi (π) molecular orbitals.
The “bonding” situation is described by constructive overlap, where p orbitals of matching phase join together to form a pi molecular orbital, which looks like this:
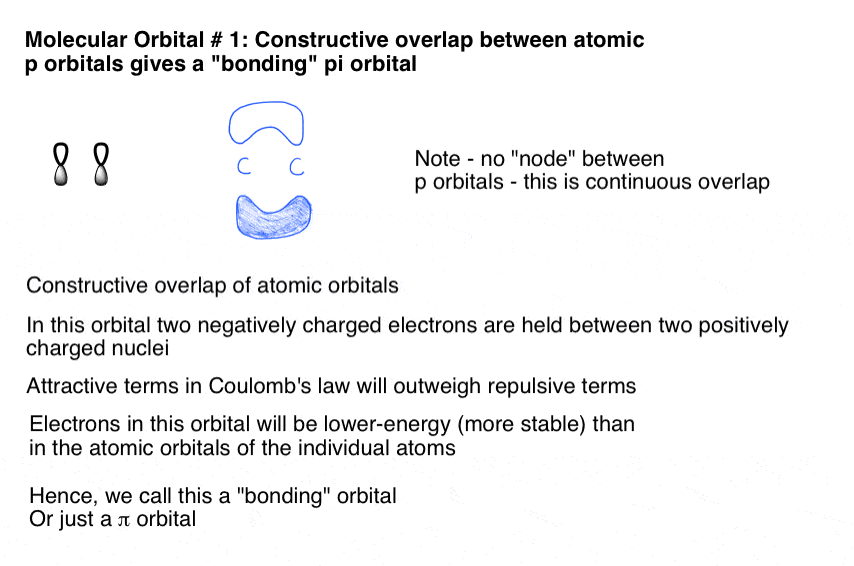
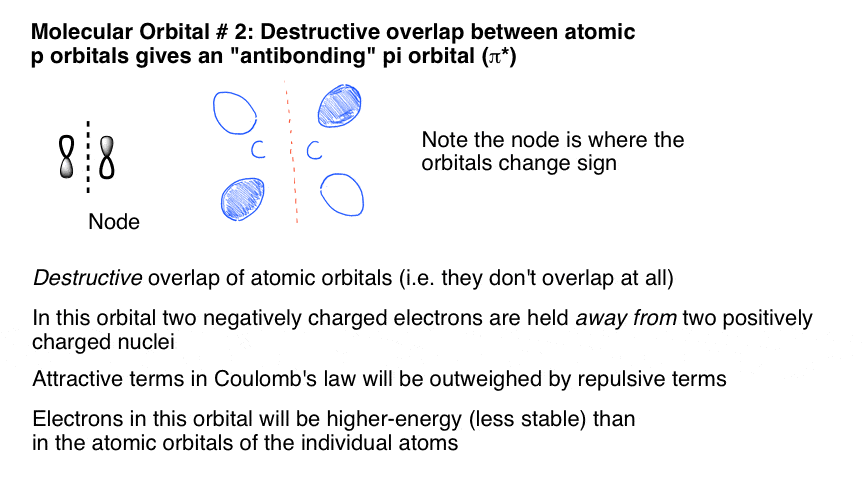
8. Antibonding Pi Molecular Orbitals Result From the Destructive Side-On Overlap Of Adjacent p Orbitals
The second molecular orbital is described by destructive overlap (or destructive interference, if you prefer) where p orbitals of mismatched phase join together to form another pi molecular orbital.
Note that this results in a node between the atoms – an area where there is a change in phase and thus no orbital overlap (and therefore no electron density). This corresponds to the “antibonding” situation we described earlier, where the two nuclei are held closely together in space but lack the attractive interactions of electrons between them. Hence it is higher in energy.
{kind=link}
9. An Energy Diagram For A Simple Pi Bond Incorporating Bonding and Antibonding Pi Molecular Orbitals
By analogy to our previous examples, we can draw up an energy diagram for these two situations. (The energy of the antibonding orbital is a little bit more than 2 times the bonding energy). [Note 3]
According to the Aufbau principle, these orbitals will fill up in order of stability, which means that for a typical pi bond, we end up with two electrons in the Pi orbital and zero in the Pi*.
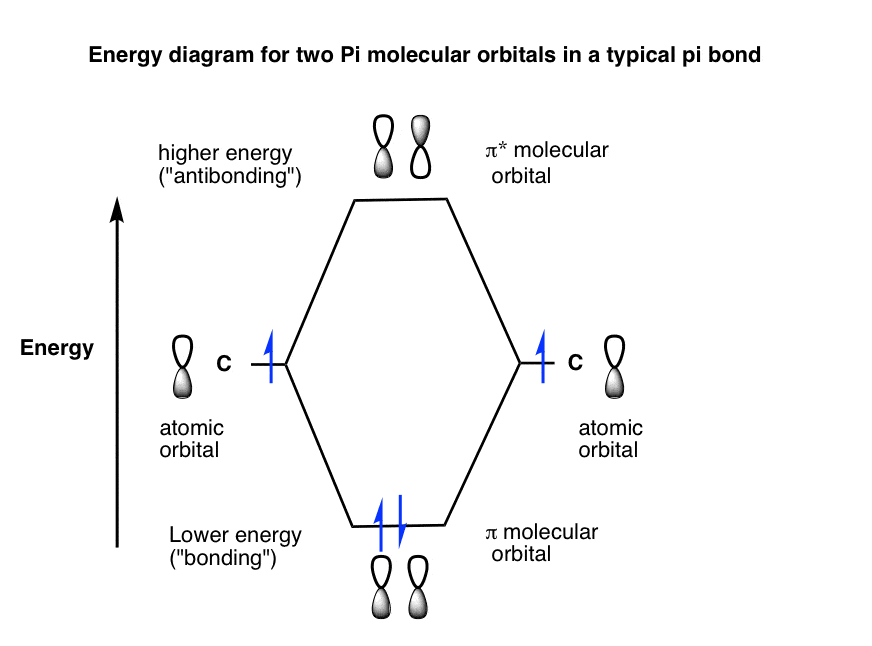
If we were to add a third electron, it must go to the Pi* (antibonding) orbital.
For the simple case of a pi bond, note that the Pi molecular orbital is the “highest occupied molecular orbital” (or HOMO) and the Pi* orbital is the “lowest-unoccupied molecular orbital”, or LUMO. This isn’t an important distinction at the moment, but it will come up in subsequent posts.
10. Conclusion: Some Rules for Pi Molecular Orbitals
Hopefully most of this was a review from first-semester material. We are going to use this exercise to form a set of guidelines that we can apply building up more complicated systems molecular orbitals that comprise three, four, or even more individual components.
Let’s review the main points.
- When overlap between adjacent p orbitals is possible, then Pi (π) molecular orbitals can be formed. [Note 4]
- The number of Pi molecular orbitals formed by overlap will always equal the number of contributing p orbitals
(Another way to say this: overlap of N atomic orbitals gives rise to N molecular orbitals) - When N=2, this describes a single Pi bond.
- A node is where phases change sign.
- The lowest-energy (most stable) orbital has zero nodes between the individual orbitals (e.g. our Pi bonding orbital)
- The highest-energy (least stable) orbital has N-1 nodes between the individual orbitals. (For our pi-bond example, since N=2, it had one node) [
In the next post we will cover systems of three and four molecular orbitals.
N=3 describes allyl systems. We will cover the molecular orbitals of the allyl cation, allyl radical, and allyl anion. If there’s time, we might even get to N=4 (butadienyl) systems too.
Before we do, a quiz:
- how many molecular orbitals will be in the allyl system?
- how many nodes will the highest-energy molecular orbital have?
- how many molecular orbitals will be in the butadienyl system?
- how many nodes will the highest energy molecular orbital have?
[answers]
Thanks to Thomas Struble for help with this post.
Notes
Note 1. This is the essence of the Born Oppenheimer approximation, which states that the motion of atoms and electrons can be separated. If you want to continue beating our relationship analogy to death, it’s similar to how even happy couples can have spats and then make up before the slow-grinding wheels of the divorce attorneys have time to operate.
Note 2. For two identical atoms the energies of the two contributing atomic orbitals are the same. As we go across the periodic table (increasing electronegativity) the energy of the atomic orbitals decreases, another way of saying they are more stable.
Note 3. The pi* is more antibonding than the pi is bonding, due to electronic repulsion. See Fleming, “Frontier Orbitals and Organic Chemical Reactions” (chapter 2) for the clearest and best treatment of molecular orbitals in organic chemistry.
Note 4. For an example of adjacent p-orbitals which do not lead to molecular orbitals, see the “bridgehead olefin” below.
This is an example of two adjacent p-orbitals which cannot form a pi bond, such as in a bridgehead olefin. The system behaves like two individual radicals.
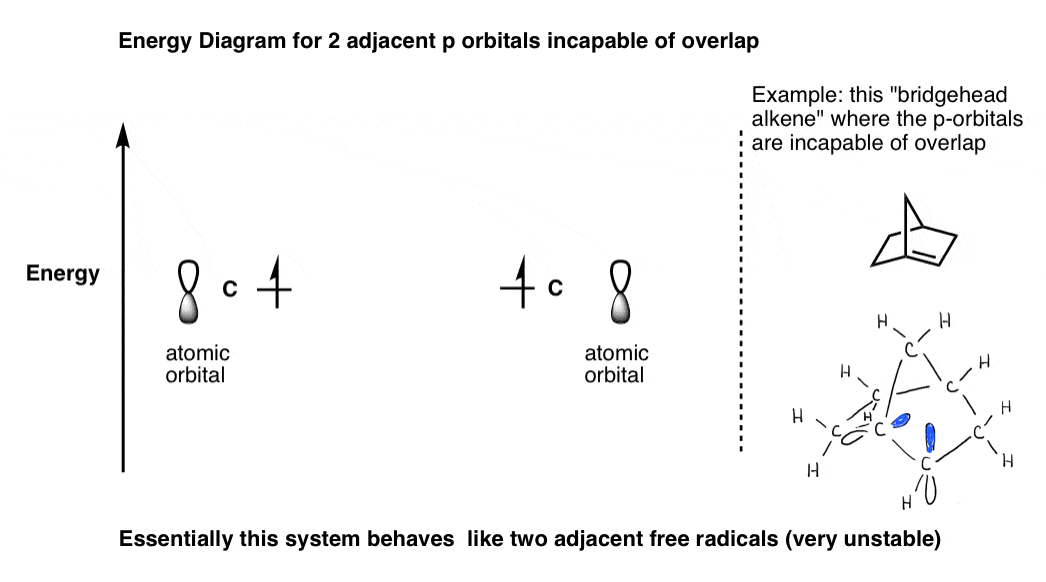
Note 5. Allyl (N=3): three p orbitals gives rise to three molecular orbitals. Highest-energy molecular orbital will have 2 nodes (N-1)
Butadienyl (N=4): four p orbitals gives rise to four molecular orbitals. Highest energy molecular orbital will have 3 nodes (N-1).
Quiz Yourself!
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.

Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.

Become a MOC member to see the clickable quiz with answers on the back.

Become a MOC member to see the clickable quiz with answers on the back.
WHAT iS MEANT by phase of an orbital ?
Hi would like to cite some of the photos in my PhD thesis how can i apply for permission?
Just cite the URL
James, this is a really excellent post by you — I thoroughly enjoyed the single/couples analogy…
In contrast what Volodymyr wrote is mind numbing and aimed way above the level of your site’s intended audience — it is an excellent example of the type of ‘pedagogy’ and scientific writing those who come here seek to avoid! Personally, I’d remove it…
I’d never know I could have such a blast while reading about orbitals. Thanks for the cheerful experience in the middle of exams!
Glad it wasn’t completely unentertaining!
Benzene Quant (Thesis).
See picture (Benzene Quant, DOI: 10.13140/RG.2.2.33278.64328),
https://archive.org/details/BenzeneQuant,
http://vixra.org/pdf/1902.0030v2.pdf
It a good visualization (Benzene Quant) of the boiling of electron-positron pairs, which always occurs in chemical bonding with any multiplicity. We got a beautiful “quantum-mechanical pattern” of a benzene molecule with a three-electron bond. I really like :), because it really conveys the meaning …
“…Therefore, in the field of chemical bonding, in this case, an electron can not be regarded as a “point object”, since it (an electron) will spend part of its time in the state “electron + pair (positron + electron)”, and therefore its interaction should be described in the framework of quantum field theory. This approach makes it possible to explain how, in the case of many-electron chemical bonds (twoelectron, three-electron, etc.), repulsion between electrons is overcome: since the chemical bond is actually a “boiling mass” of electrons and positrons, virtual positrons “help” overcome the repulsion between electrons. This approach assumes that the chemical bond is in fact a closed spatial bag (a potential well in the energy sense), in which “boiling” of real electrons and also virtual positrons and electrons occurs, and the “volume” of this potential bag is actually a “volume” of chemical bond and also the spatial measure of the quantum-mechanical uncertainty in the position of the electron.
Strictly speaking, with such a consideration, the electron no longer has a certain energy, momentum, coordinates, and is no longer a “point particle”, but actually takes up the “whole volume” of chemical bonding. It can be argued that in the chemical bond a single electron is depersonalized and loses its individuality, in fact it does not exist, but there is a “boiling mass” of real electrons and virtual positrons and electrons that by fluctuate change each other. That is, the chemical bond is
actually a separate particle, as already mentioned, a semivirtual particle. Moreover, this approach can be extended to the structure of elementary particles such as an electron or a positron: an elementary particle in this consideration is a fluctuating vacuum closed in a certain spatial bag, which is a potential well for these fluctuations.
It is especially worth noting that in this consideration, electrons are strongly interacting particles, and therefore the Pauli principle is not applicable to chemical bond (for more details see section “The Pauli Principle and the Chemical Bond”) and does not prohibit the existence of the same threeelectron bonds with a multiplicity of 1.5.”
pp. 101 – 103 Review. Benzene on the Basis of the Three-Electron Bond. (The Pauli Exclusion Principle, Heisenberg’s Uncertainty Principle and Chemical Bond).
http://vixra.org/pdf/1710.0326v4.pdf https://dx.doi.org/10.2139/ssrn.3065288
Benzene on the basis of the three-electron bond:
Review (138 pages, full version). Benzene on the Basis of the Three-Electron Bond. (The Pauli exclusion principle, Heisenberg’s uncertainty principle and chemical bond). http://vixra.org/pdf/1710.0326v4.pdf https://dx.doi.org/10.2139/ssrn.3065288
The present work shows:
1.The aromatic bond is a three-electron bond in flat cyclic systems with a specific interaction of electrons through the cycle.
2.The present work shows the inapplicability of the Pauli principle to the chemical bond (pp. 103-105).
3.In addition, a new theoretical model of the chemical bond is proposed on the Heisenberg uncertainty principle (pp. 92-103).
4.That is, in fact, in this paper it is shown that modern concepts of the chemical bond can not be strictly considered theoretically true, but rather qualitative with empirical quantitative calculations.
Bezverkhniy Volodymyr (viXra): http://vixra.org/author/bezverkhniy_volodymyr_dmytrovych
Volodymyr Bezverkhniy (SSRN): https://papers.ssrn.com/sol3/cf_dev/AbsByAuth.cfm?per_id=2828345
Volodymyr Bezverkhniy (Quora): https://www.quora.com/profile/Volodymyr-Bezverkhniy
Bezverkhniy Volodymyr (archive): https://archive.org/details/@threeelectronbond
You can post this stuff here, Volodymyr, but that doesn’t mean that it’s the best place for it.
Quantum-mechanical analysis of the MO method and VB method from the position of PQS.
http://vixra.org/pdf/1704.0068v1.pdf
The MO method and the VB method are analyzed using the principle of quantum superposition (PQS) and the method of describing a quantum system consisting of several parts. It is shown that the main assumption of the molecular orbitals method (namely, that the molecular orbital can be represented like a linear combination of overlapping atomic orbitals) enters into an insurmountable contradiction with the principle of quantum superposition. It is also shown that the description of a quantum system consisting of several parts (adopted in quantum mechanics) actually prohibits ascribe in VB method to members of equation corresponding canonical structures.
Using the quantum superposition principle, the MO method and the VB method were analyzed and it is shown that they are in contradiction with quantum mechanics.
Also, using the quantum-mechanical description of a system consisting of several parts, it is shown that the attribution of canonical structures to the members of the equation is incorrect. Therefore, both the MO method and the VB method did not describe molecules with chemical bonds but actually, a lot of atoms (of which the described molecules consisted). That is, in the quantum chemical calculations, the chemical bond was “lost”. Therefore, in order to “introduce” a chemical bond into calculations and avoid conflict with quantum mechanics, it is suggested to postulate the existence of MO as a new fundamental quality that describes a specific chemical bond and is not derived from simpler structural elements.
Quantum-mechanical analysis of the MO method and VB method from the position of PQS:
http://vixra.org/pdf/1704.0068v1.pdf
1. Structure of the benzene molecule on the basis of the three-electron bond.
http://vixra.org/pdf/1606.0152v1.pdf
2. Experimental confirmation of the existence of the three-electron bond and theoretical basis ot its existence.
http://vixra.org/pdf/1606.0151v2.pdf
3. A short analysis of chemical bonds.
http://vixra.org/pdf/1606.0149v2.pdf
4. Supplement to the theoretical justification of existence of the three-electron bond.
http://vixra.org/pdf/1606.0150v2.pdf
5. Theory of three-electrone bond in the four works with brief comments.
http://vixra.org/pdf/1607.0022v2.pdf
6. REVIEW. Benzene on the basis of the three-electron bond (full version, 93 p.).
http://vixra.org/pdf/1612.0018v5.pdf
7. Quantum-mechanical aspects of the L. Pauling’s resonance theory.
http://vixra.org/pdf/1702.0333v2.pdf
8. Quantum-mechanical analysis of the MO method and VB method from the position of PQS.
http://vixra.org/pdf/1704.0068v1.pdf
Bezverkhniy Volodymyr (viXra): http://vixra.org/author/bezverkhniy_volodymyr_dmytrovych
Bezverkhniy Volodymyr (Scribd):
https://www.scribd.com/user/289277020/Bezverkhniy-Volodymyr#
Bezverkhniy Volodymyr (Amazon): https://www.amazon.com/Volodymyr-Bezverkhniy/e/B01I41EHHS/ref=dp_byline_cont_ebooks_1
This screenshots (foto) (most with explanation) see by this link.
Bezverkhniy Volodymyr (Archive.org):
https://archive.org/details/@threeelectronbond
Sincerely Bezverhny Volodymyr Dmitrievich.
My ORCID iD: 0000-0002-3725-5571