Alkene Reactions
Stereoselective and Stereospecific Reactions
Last updated: May 28th, 2026 |
Stereoselective and Stereospecific Reactions
An ideal chemical reaction would be selective for one product only. This would mean no time spent purifying our product and no wasted materials.
In the real world, reactions give mixtures. Not only can reactions occur with different functional groups on a given molecule, but even reactions on a single functional group can give rise to various types of isomers.
We can classify the different types of selectivity in organic chemistry into several important categories.
- Chemoselective reactions are selective for one functional group over another – for example, a reaction that selectively reacts with alkynes in the presence of alkenes.
- Regioselective reactions are selective for the formation of one constitutional isomer over another – for example, the “Markovnikov-selective” addition of strong acids to alkenes is an example of regioselectivity, as is “Zaitsev’s rule” for elimination reactions.
- Stereoselective reactions are selective for the formation of one stereoisomer over another, such as the preference for partial hydrogenation of alkynes with Na/NH3 to give trans alkenes.
Whether or not a given combination of starting material and reactant is regioselective or stereoselective depends on the reaction mechanism, as well as on the specific structure of the starting material.
Selectivity implies some value less than perfection. A reaction which gives a 99:1 ratio of products is highly selective, but we still use the word “selective”.
There are some reactions which are so stereoselective that they give one stereoisomer exclusively. This is often due to the existence of a concerted reaction mechanism (like the SN2). The the term stereospecific is used to refer to reactions where starting materials differing in configuration at a single carbon (i.e. stereoisomers) result in products that are also stereoisomers.
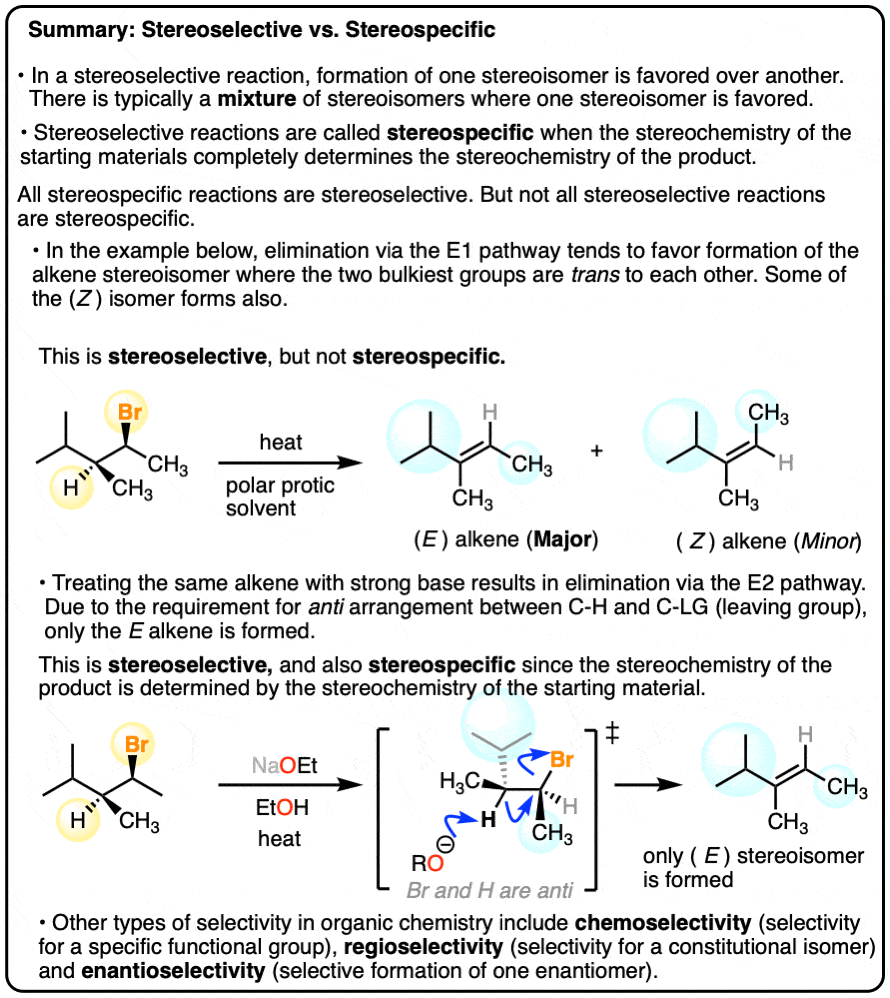
Table of Contents
-
- Selectivity Is A Highly Desirable Property For A Reaction To Have
- The Different Classes of “Selectivity”
- Chemoselectivity: Functional Group Selectivity
- Regioselectivity
- Stereoselective vs. Non-Stereoselective Reactions
- Stereoselective vs. Stereospecific Reactions
- Enantioselectivity
- Diastereoselectivity
- The Substrate Has A Vote On Whether A Reaction Is Selective
- Notes
- Quiz Yourself!
- (Advanced) References and Further Reading
1. Selectivity Is A Highly Desirable Property For A Reaction To Have
Let’s face it. We’re accustomed to things that have a single output for every input.
I mean, when you push the “L” button on a keyboard, you’re used to seeing an “L” show up on the screen 100% of the time. If that doesn’t happen, then someone hasn’t done their job.
So imagine what it would be like to push the “L” button on your keyboard a hundred times and getting 74 L’s, 22 K’s, and various amounts of J, R, S, and T. That would be kind of annoying, right? Well…. welcome to life in the organic chemistry lab.
For instance, imagine that we run a reaction with two reactants, A and B. In an ideal world, we’d get a 100% yield of product C.
But that’s not generally how organic chemistry reactions work. Instead, in this example, we get a mixture of products C, D, and a little bit of E.
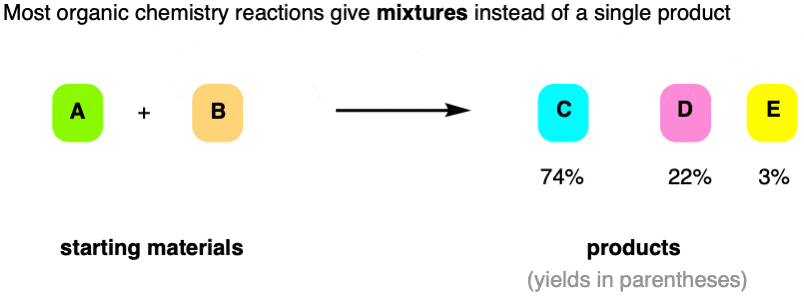
And hey, there are many contexts in which a 74% yield is actually considered “good”!
If your reaction produces a 60% yield of C, 30% D, and 10% E, you can reasonably say that the reaction is selective for C. Maybe not “highly selective”, but at least “somewhat selective”.
On the other hand, a reaction that gave a 33% yield each of C, D, and E can be safely called “unselective”. [Note 1]
Calling a reaction “selective” is a bit of a judgement call. You could make a case that a reaction that reproducibly gives a 55:45 mixture of products is selective, but you’d have to admit the selectivity is poor. On the other hand, a reaction that gives a 200:1 ratio of products (at the limits of detection for practical purposes) can clearly be called “highly selective”.
Selectivity is a highly desirable property for a reaction to have.
Organic chemists go to great lengths to maximize the selectivity of reactions, since it means less time spent on purification and less money spent on wasted reactants that give unwanted byproducts we need to dispose of.
Numerous Nobel Prizes have been given to the discoverers of reactions that are highly selective in some way – the 2022 Nobel for “click chemistry” being the most recent. [Note 2] You might not appreciate it now, but when you take an organic chemistry course, we’re really trying to teach you some of the best and most selective reactions that are known to us!
On the other hand, there is a large class of reactions in organic chemistry that result in the formation of thick black tar on the bottom of the reaction vessel. These reactions firmly belong in the “unselective” category.
2. The Different Classes of “Selectivity”
It turns out that there are lots of different ways in which a reaction can be unselective, and therefore many opportunities for the formation of multiple products!
- The first reason is that many reagents can react with several different functional groups on a molecule, giving rise to completely different products.
- The second reason is that even if a given reagent reacts only with one functional group, the reaction can still result in the formation of various types of isomers, such as constitutional isomers (also known as “regioisomers” in this context), and stereoisomers. (See article: Types of Isomers).
The most important factor in ensuring a selective reaction is to simply to choose a substrate (also known as “starting material”) with a minimum of reactive functional groups and possibilities for isomerism. (For more on possibilities of isomerism, including quizzes, “What’s a Racemic Mixture“).
The selectivity of a reaction will also strongly depend on the reaction mechanism, and to a lesser extent on some external factors such as temperature and solvent.
The word, “selectivity” in organic chemistry can be applied to several different situations. It can be helpful to start by thinking, “selective for what?”.
For our purposes, we’ll generally discuss 4 types of selectivity:
- A chemoselective reaction is selective for the reaction at one functional group over another functional group.
- A regioselective reaction is selective for the formation of one constitutional isomer (“regioisomer”) over another
- A stereoselective reaction is selective for the formation of one stereoisomer over another.
- An enantioselective reaction is selective for the formation of one enantiomer over another.
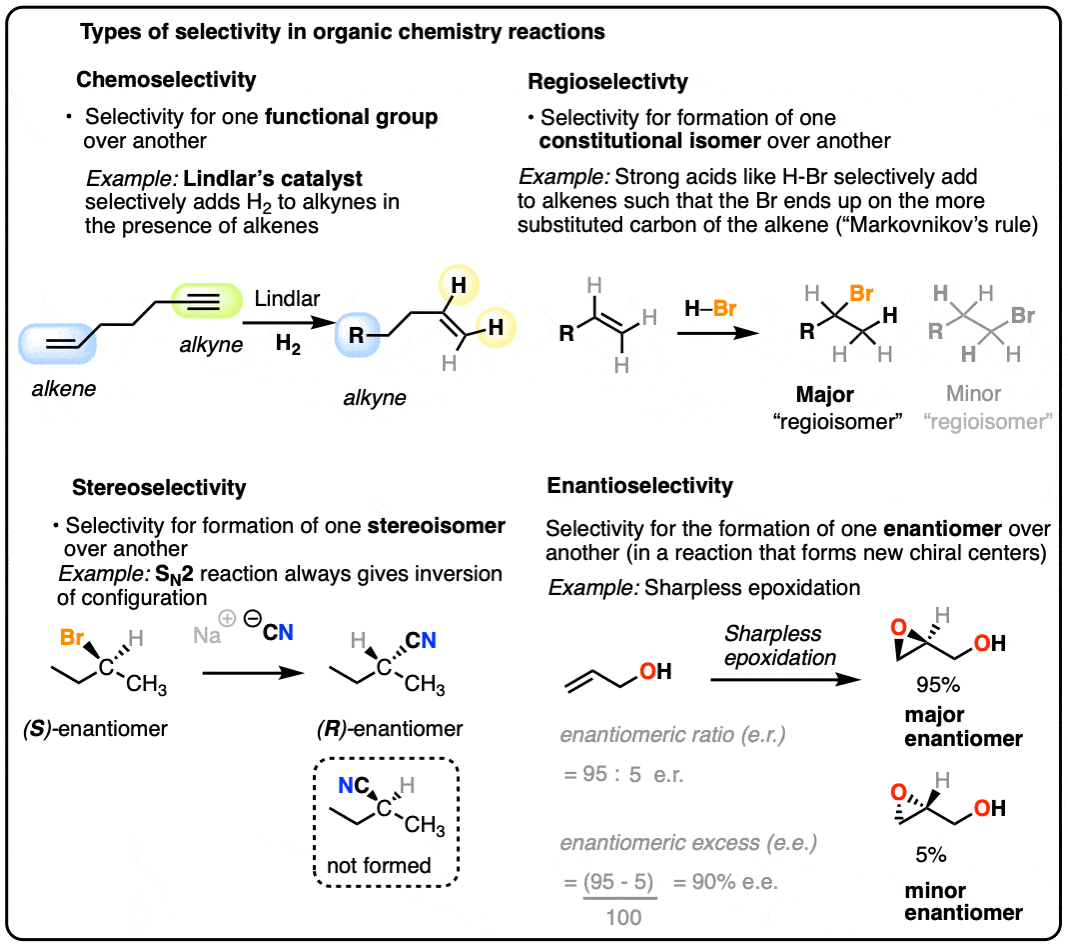
Note that these types of selectivity are not mutually exclusive, and multiple classes of selectivity can operate at the same time.
The E2 reaction, for example, is regioselective (for the more substituted “Zaitsev” alkene) as well as stereoselective (for anti-elimination) but not enantioselective (since no chiral centers are formed). (See article: The E2 Reaction)
3. Chemoselectivity
Any time a given reaction is selective for a certain functional group, that’s an example of “chemoselectivity”.
The term “chemoselectivity” doesn’t come up too much in introductory organic, mostly because for simplicity’s sake you’re often only shown molecules that have a single reactive functional group (alkanes and ethers are generally not considered “reactive” functional groups for most purposes).
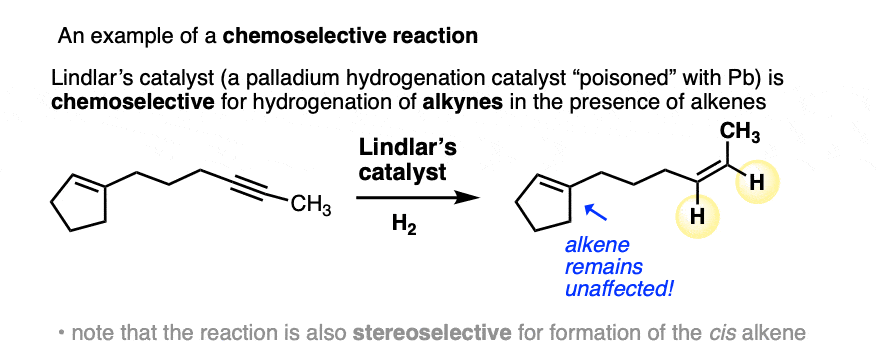
There are some examples in introductory organic where you are explicitly told that a given reagent is selective for one functional group over another. For example, in the chapter on alkynes, Lindlar’s catalyst is selective for the hydrogenation of alkynes over alkenes. (See article: Lindlar’s Catalyst for Partial Hydrogenation of Alkynes). That’s a classic example of chemoselectivity.
Another case that comes up frequently is when a given reagent is capable of acting as both a base and a nucleophile on a given substrate, which can lead to multiple products. (See article: Basicity vs Nucleophilicity)
Alkyl halides are a perfect example. If you’ve ever been asked to decide if an alkyl halide goes through an SN2 or E2 reaction with a given nucleophile/base, that’s ultimately a chemoselectivity question.
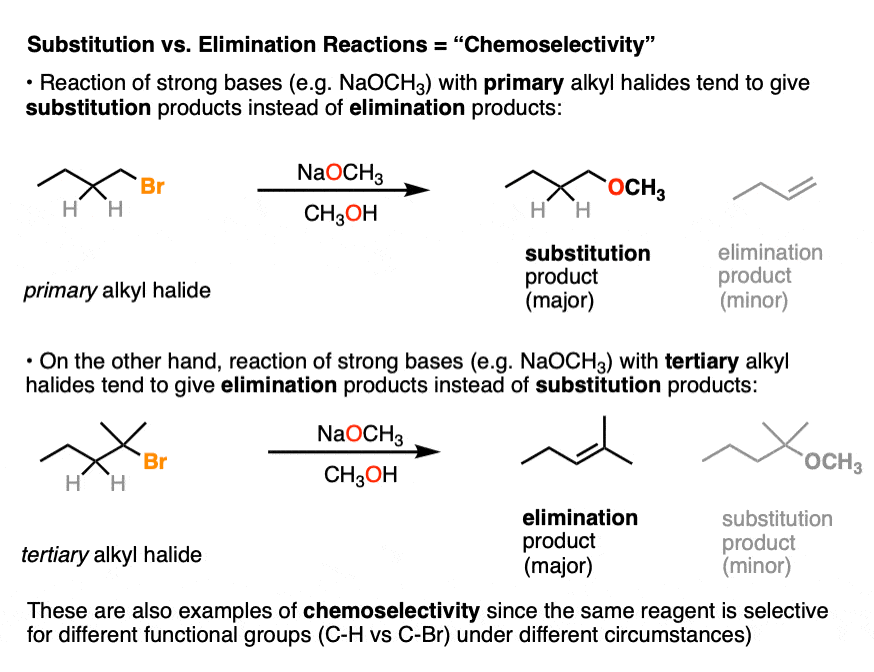
Because in introductory organic chemistry we mostly only deal with molecules containing a single functional group, I’m not going to drill down much further on chemoselectivity, although further along in the course, you’ll see examples of protecting groups as one way of trying to solve the problems of chemoselectivity. (See article: Protecting Groups for Alcohols)
(Other examples of chemoselective reactions: selective reduction of aldehydes/ketones over esters by NaBH4; selective deprotection of amine protecting groups in peptides; )
4. Regioselectivity
Even if a reaction is confined to a single functional group, the formation of isomers is an extremely common result.
Constitutional isomers are molecules with the same empirical formula, but different connectivity.
A classic example of a reaction that forms constitutional isomers is in the addition of strong acids to alkenes (e.g. HCl, HBr, HI, and H3O+). For example, reaction of propene with hydrochloric acid (HCl) results in a mixture of 2-chloropropane and 1-chloropropane, both with the empirical formula C3H7Cl.
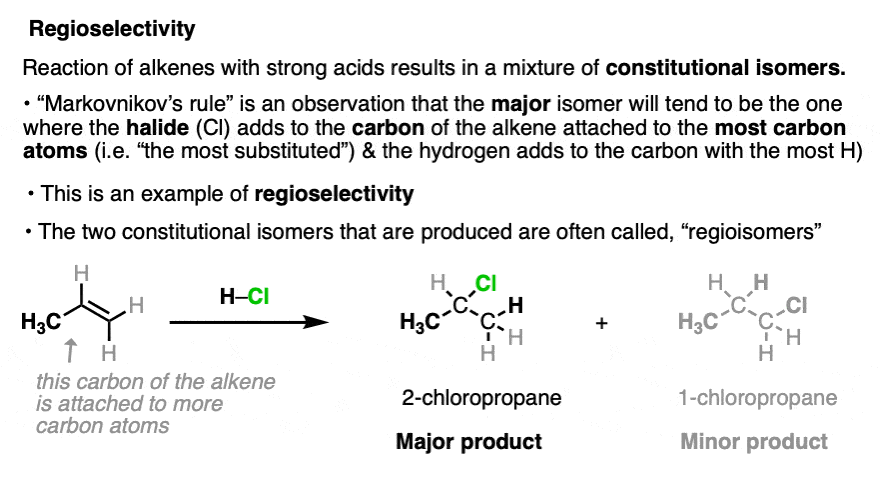
In these reactions it is observed that the major product is the one where the C-Cl bond forms on the carbon of the alkene directly attached to the most carbon atoms (“most substituted”), and the C-H bond forms on the carbon of the alkene directly attached to the fewest carbon atoms (the “least substituted” carbon). This is known as “Markovnikov’s Rule” (See article: Hydrohalogenation of Alkenes and Markovnikov’s Rule).
This type of selectivity is known as “regioselectivity“, (from regio-, Latin for “region”) and the constitutionally isomeric products are often referred to as regioisomers. [Note 3]
In the case of addition of acids to alkenes, this regioselectivity arises due to the greater stability of carbocation intermediates on more substituted carbons (See article: Carbocation Stability).
This brings us to a subtle point.
The regioselectivity (and stereoselectivity, for that matter) of a reaction is highly dependent on the properties of the reaction mechanism itself. But the mechanism is not the only factor.
Selectivity is also a function of the structure of the substrate (i.e. starting material).
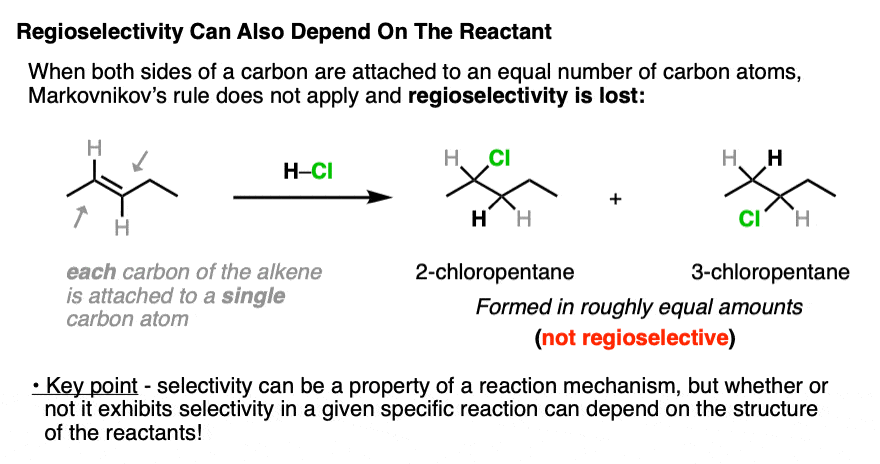
Markovnikov’s rule for regioselectivity only holds when one terminus of the alkene is bonded to more carbons than the other. If they are bonded to an equal number of carbons, then the carbocations derived from those alkenes will be roughly equal in stability, and the regioselectivity will go away.
When each carbon of the alkene is directly connected to the same number of carbon atoms, regioselectivity is lost.
Another example of regioselectivity in organic chemistry is Zaitsev’s Rule in the elimination of alkenes, where formation of the more substituted alkene tends to be more favored. (See article: Elimination Reactions – Zaitsev’s Rule).
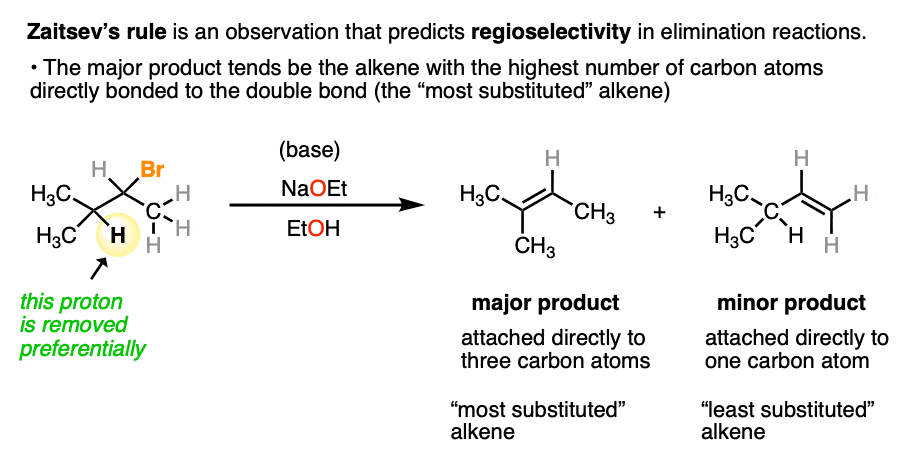
This reaction tends to be regioselective due to the greater stability of alkenes as C-H bonds are replaced with C-C bonds. (See article: Alkene Stability)
Furthermore, not all reactions are capable of forming constitutional isomers (regioisomers). So not all reactions have the possibility of being regioselective.
For example, when two identical bonds are formed in an addition to an alkene or alkyne, there is no possibility of forming constitutional isomers. Hydrogenation and dihydroxylation are two examples of reactions for which the concept of “regioselectivity” does not apply. (others include epoxidation, cyclopropanation, and halogenation).
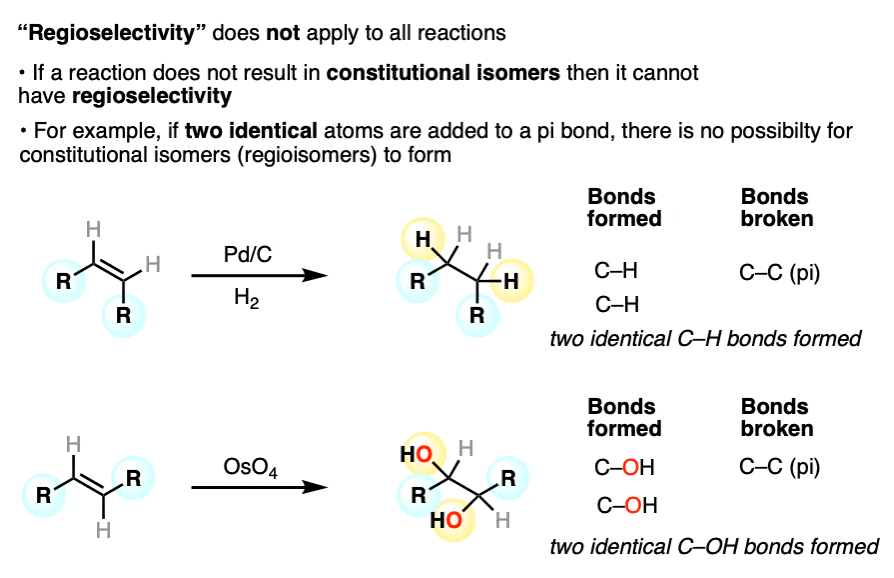
Other examples of regioselective reactions: ortho-,para- vs meta selectivity in electrophilic aromatic substitution; Diels-Alder reactions; 1,4-additions of Gilman reagents; radical addition of HBr to alkenes; hydroboration of alkenes; free-radical substitution of tertiary C-H bonds with Br2)
5. Stereoselectivity
When a reaction has the possibility of forming stereoisomers, the issue of stereoselectivity arises.
This article assumes you are familiar with the difference between diastereomers and enantiomers. For more clarification, see Types of Isomers.
Reactions that proceed through carbocation or radical intermediates often, (but not always), result in poor stereoselectivity.
As we’ve seen, the addition of acids to alkenes (we discussed above) is certainly regioselective with certain alkenes. However, the stereoselectivity of the reaction is poor. [Note 4]
When we add H-Br to the alkene below (1,2-dimethylcyclohexene), a roughly equal mixture of cis- and trans– stereoisomers are obtained.
(this is due to the formation of a trigonal planar carbocation intermediate, which can be attacked equally well on either face by the halide ion).
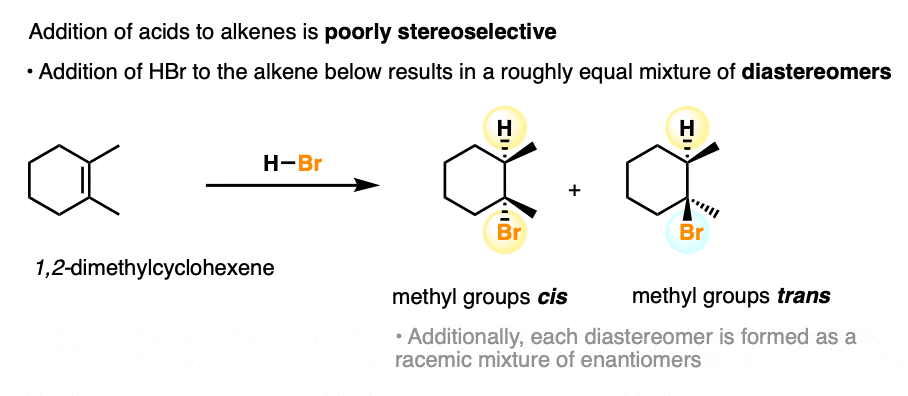
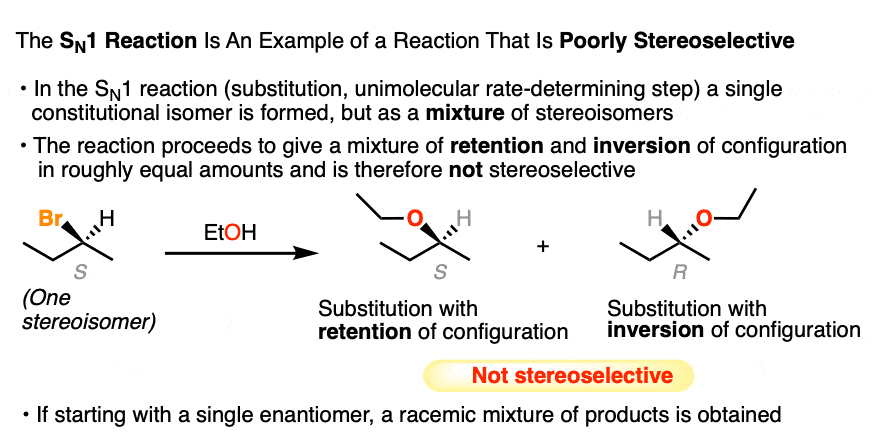
Another classic example of a poorly stereoselective reaction is the unimolecular substitution reaction (SN1) of alkyl halides, where we can start with a single pure enantiomer and end up with a racemic mixture of products (a racemic mixture is a 1:1 mixture of enantiomers).
Like addition of H-Br to alkenes, the SN1 also proceeds through a carbocation intermediate that can be attacked with equal likelihood from either face, giving us a mixture of products representing retention and inversion of stereochemistry (See article – the SN1 Mechanism). [Note 4]
Some reactions the proceed through carbocation intermediates can still have relatively high stereoselectivity.
In the E1 reaction, a carbocation intermediate undergoes loss of a proton to form an alkene as the final product. Elimination will generally favor formation of the alkene where the bulkiest groups are trans to each other since this minimizes steric strain. (See article – Alkene Stability).
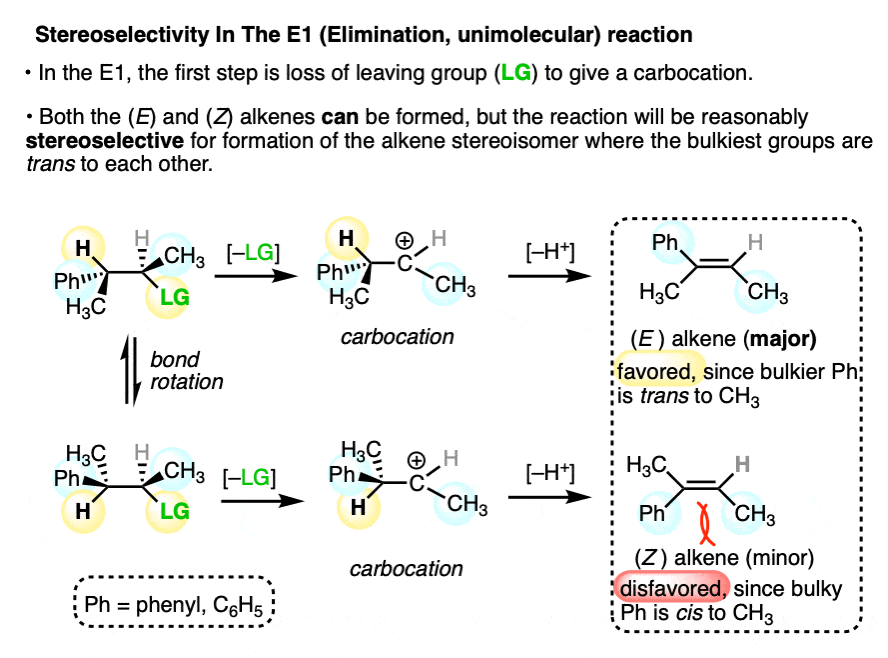
Since the (E) and (Z) alkenes are stereoisomers of each other, and the reaction favors the (E), this is an example of stereoselectivity.
More examples of stereoselective reactions – partial hydrogenation of alkynes (both Lindlar and Na/NH3), hydroboration of alkenes, the Diels-Alder reaction, Cope rearrangement.
6. Stereoselective vs. Stereospecific Reactions
If a reaction gives a 99:1 mixture of stereoisomers, we can feel pretty confident in calling that reaction, highly”stereoselective”.
However, some reactions are so spectacularly stereoselective for the formation of a single type of product that the term “stereoselective” doesn’t seem to do it proper justice.
For example, dihydroxylation of alkenes gives only addition products where the two C-OH bonds form on the same face of the alkene. Absolutely none of the addition products are formed where the two C-OH bonds are trans.
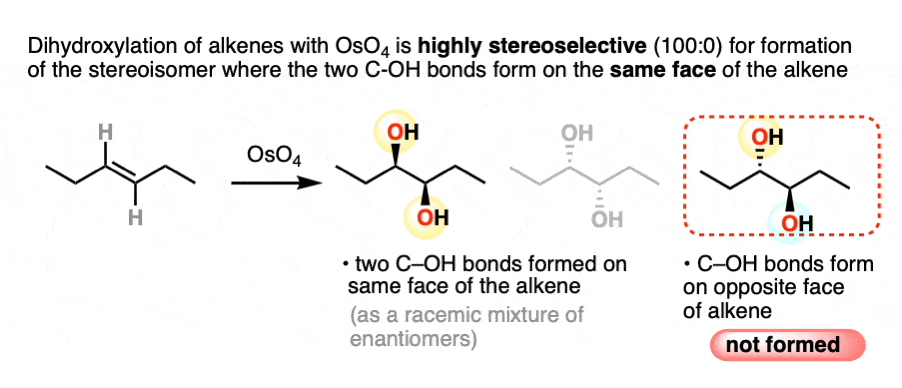
Sometimes, the term stereospecific is used to categorize reactions like this – reactions that are so super-dee-duper stereoselective that a given stereoisomer is formed exclusively.
IUPAC (which you can think of as the Ministry of Magic, but for chemistry) frowns upon this use of the word stereospecific.
According to IUPAC, a stereospecific reaction is one where starting materials differing only in their configuration are converted into stereoisomeric products.
The SN2 reaction is an example of a stereospecific reaction. Carrying out the SN2 on starting materials that are enantiomers of each other results in products that are themselves enantiomers of each other! (See article – The SN2 Mechanism).
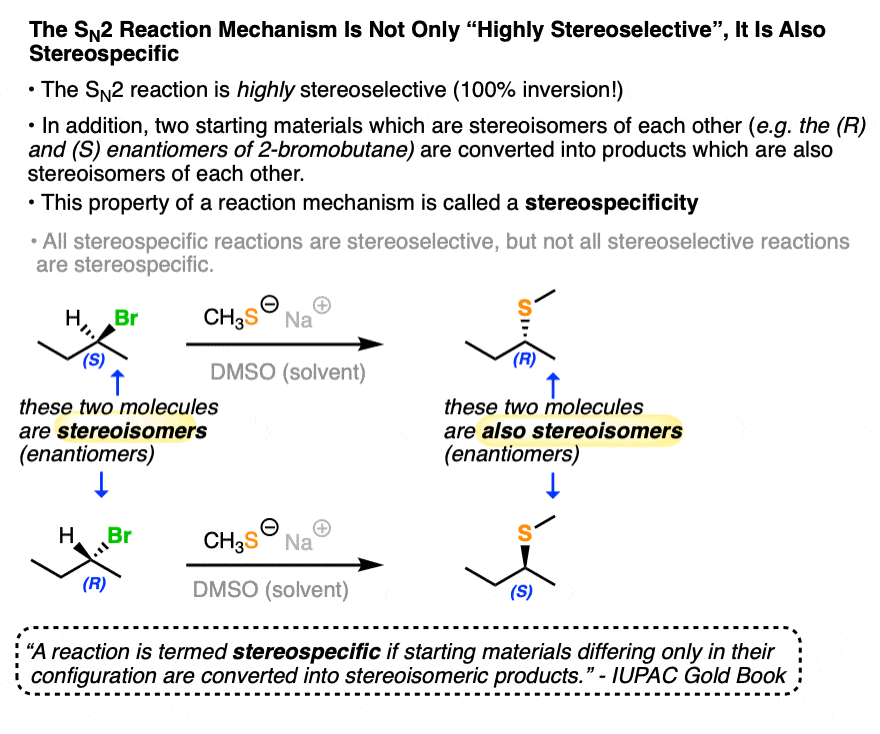
Compare that to the SN1 reaction, above, which is poorly stereoselective.
A slightly preferable definition (thx, Clayden) is that in a stereospecific reaction, the stereochemistry of the starting material determines the stereochemistry of the product.
This generally points to a concerted mechanism where only one stereochemical outcome is possible. We can say that the reaction doesn’t have a choice as to which product is made.
In the E1 reaction we saw above, two alkene stereoisomers were formed. If the same starting material is treated with a strong base, the E2 mechanism (elimination, bimolecular rate-determining step) occurs instead, and we get one stereoisomer exclusively.
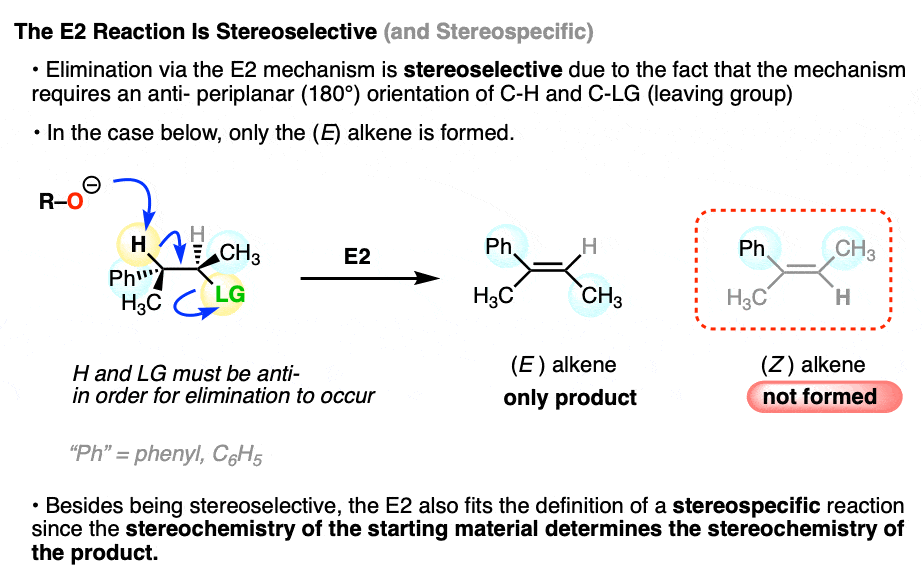
This is why the E2 gets far more use in modern organic chemistry than the E1. Since the E2 is stereospecific, you have much more control over the stereochemistry of the product and are much less likely to get mixtures.
Halogenation of alkenes is another stereospecific reaction. The (E) and (Z) isomers of a given alkene will give rise to products that are stereoisomers of each other. Another example of the stereochemistry of the starting material determining the stereochemistry of the product.
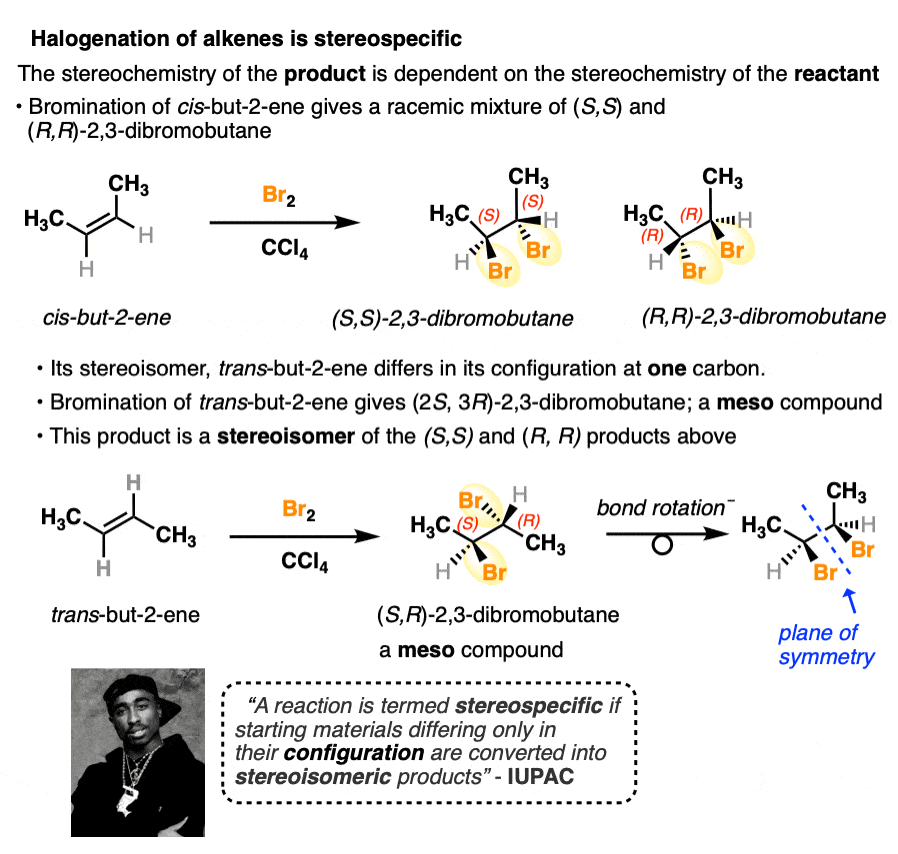
There are many more examples of stereospecific reactions we will cover in this course.
A non-exclusive list of other stereospecific reactions includes epoxidation, cyclopropanation, halogenation of alkenes, the Diels-Alder reaction, and electrocyclic reactions.
All stereospecific reactions are stereoselective, but not all stereoselective reactions are stereospecific.
A final example of a reaction that is stereoselective, but not stereospecific, is the hydrogenation of alkenes with Pd/C and H2. See article – Hydrogenation of Alkenes with Pd/C). [Note 5]
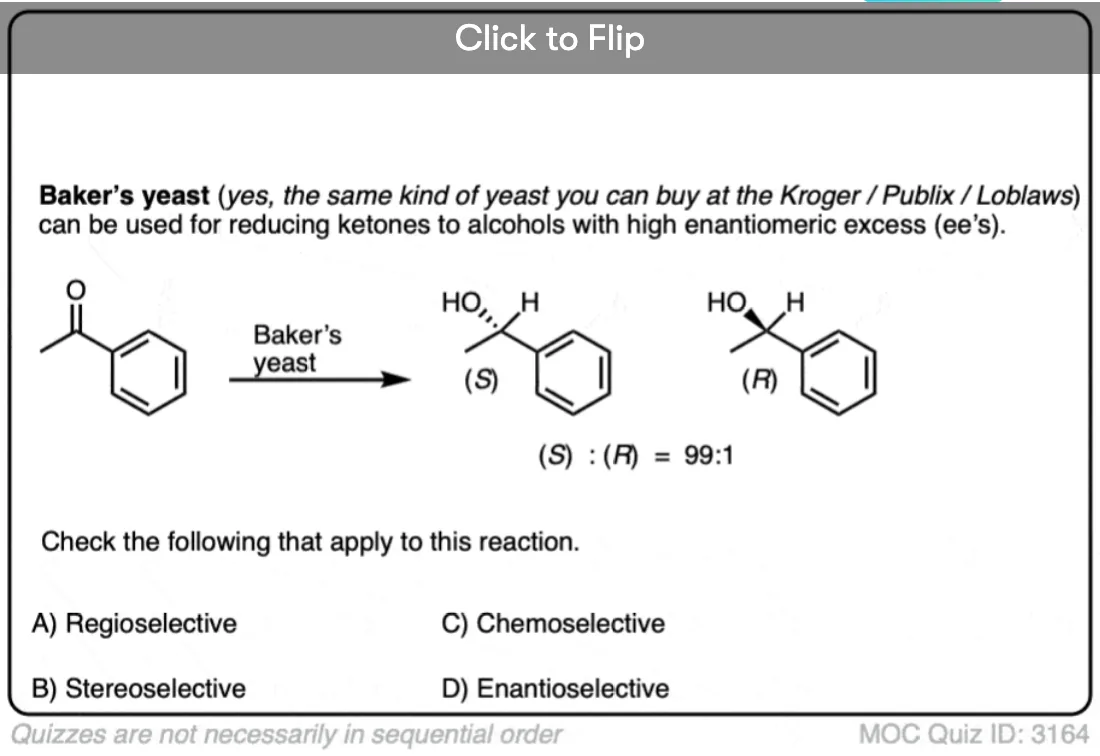
7. Enantioselectivity
A subset of stereoselective reactions are enantioselective reactions, where one enantiomer is formed selectively over another.
Enantioselectivity requires the influence of some kind of chiral material, such as a chiral reagent or starting material. (As Matt would say, “optical inactivity is preserved”).
Although many enantioselective reactions exist, we tend not to cover them so much in Org 1 / Org 2. If you take biochemistry, you will no doubt see many examples of enantioselective reactions carried out by enzymes.
One enantioselective reaction that does see some action is the enantioselective epoxidation of allylic alcohols, developed by the laboratory of Barry Sharpless (Nobel Prize in Chemistry, 2001 and 2022) and is known as the Sharpless asymmetric epoxidation.
In the Sharpless epoxidation, an allylic alcohol is treated with a catalytic amount of a titanium compound, a stoichiometric amount of oxidant (usually t-butylhydroperoxide, t-BHP) and a catalytic amount of an ester derived from one of the enantiomers of tartaric acid.
The Sharpless gives very good enantioselectivity for the epoxidation of a wide variety of allylic alcohols.
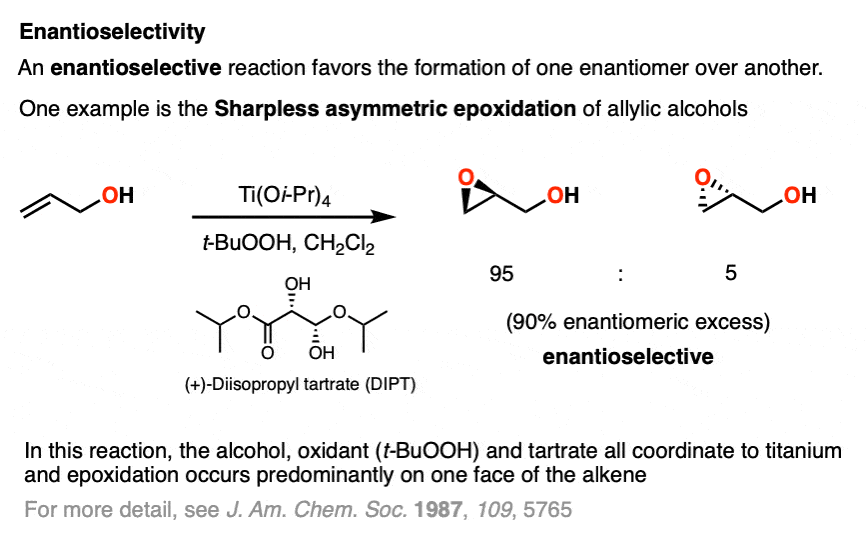
Note that enantioselectivity is measured by enantiomeric excess (e.e.) or sometimes enantiomeric ratio (e.r.) (See article – Optical Activity and Enantiomeric Excess).
Enantioselective variants of many common reactions have been developed and you will undoubtedly encounter them if you enroll in more advanced organic chemistry courses.
Enantiomeric excess = (mole fraction of major enantiomer) – (mole fraction of minor enantiomer) expressed as a percentage. A racemic mixture has an enantiomeric excess of 0 and a 99:1 ratio of enantiomers has an enantiomeric excess of 98. See: Optical Purity and Enantiomeric Excess).
8. Diastereoselectivity
Diastereomers are stereoisomers that are not mirror images of each other (i.e. stereoisomers that are not enantiomers).
A common situation where diastereomers are formed is when a molecule with a pre-existing chiral center undergoes a reaction at a functional group (e.g. an alkene).
Remember that in order for molecules to be enantiomers, the configuration must be reversed at all chiral centers (See article – How To Draw The Enantiomer of a Chiral Molecule).
Since the configuration at the pre-existing chiral center is unaffected by the epoxidation reaction, these can’t be enantiomers. They are diastereomers.
One of the most common examples of diastereoselective reactions occurs when one face of an alkene is partially “blocked” by the presence of nearby groups that hinder the approach of a reagent. In these cases the favored product will be the one where the reagent approaches from the face where it encounters less steric hindrance.
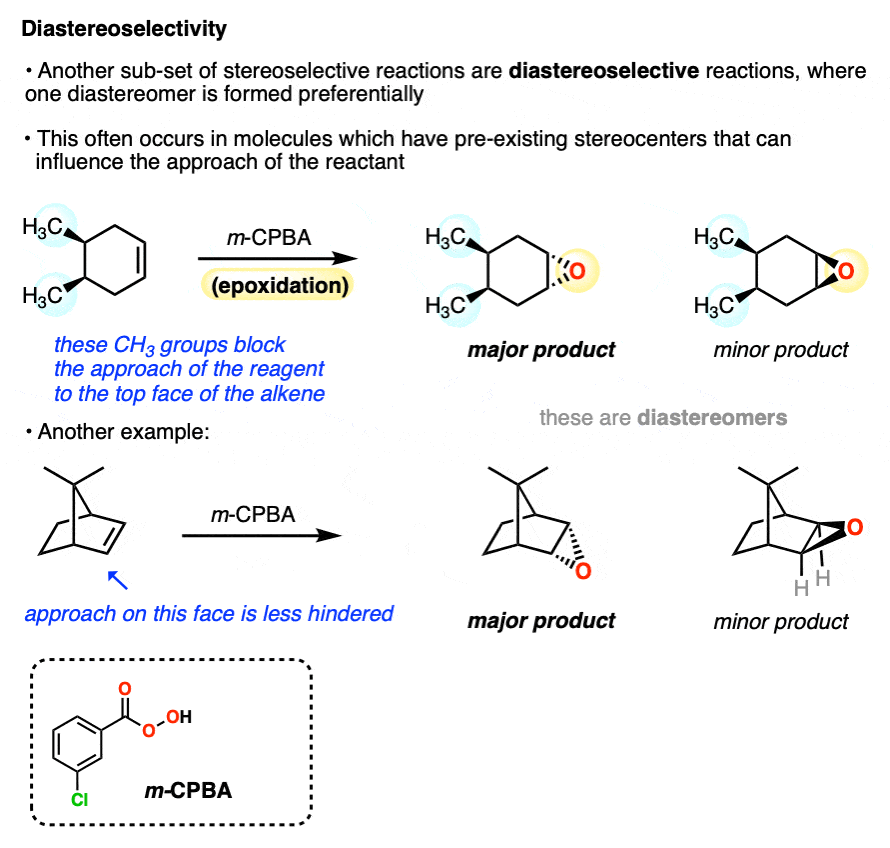
The preference for one diastereomer over another is called “diastereoselectivity”.
Endo and exo products in the Diels-Alder reaction are another prominent example of diastereoselectivity. (See article – Exo and Endo Products In the Diels-Alder).
9. The Substrate Has A Vote On Whether A Reaction Is Selective
One question that comes up a lot is, “how do I know if a reaction will lead to constitutional isomers, a racemic mixture, or a mixture of diastereomers?”
My answer to that is not very satisfying. “It depends!”
Depends on what? Two key factors.
The first factor is the pattern of bonds that form and break for that particular reaction, along with its pattern for regioselectivity (i.e. is it Markovnikov-selective? Zaitsev selective?) and its pattern for stereoselectivity.
Assuming you have that key pattern close at hand, the next step is to apply it to the specific starting material (“substrate’) in question.
Whether a reaction will form enantiomers, diastereomers, or constitutional isomers will depend on the specific structure of the substrate. There is no way to memorize your way through this. You just have to apply the patterns to the reaction at hand.
For lots of examples (with quizzes) of this type of thinking, see, “What’s A Racemic Mixture?“.
Notes
Note 1. If C-H bonds are involved, statistics should be factored in when calculating selectivity.
In the chapter on free radical reactions we covered the selectivity (or lack thereof) of free-radical chlorination reactions relative to bromination reactions. The chlorination of propane gives 55% 2-chloropropane and 45% 1-chloropropane. This is indeed not very selective, but when you consider the fact that there are 6 methyl hydrogens and 2 methylene hydrogens, a completely unselective reaction would give us a 25:75 ratio of 2-chloropropane to 1-chloropropane. See article – Selectivity in Free Radical Reactions.
Note 2.The most recent example of a Nobel Prize in Chemistry being awarded for a highly selective reaction is the 2022 Prize for “click chemistry”. One of the awardees (Bertozzi) developed a reaction that joins together two reaction partners even when they are swimming around together in a living cell.
Note 3. Another example of regioisomer formation that we won’t discuss in detail here is when two or more of the same functional groups are present on a molecule and the reagent may react with either one.
For example the molecule below contains two alkenes, and therefore two regioisomers may form. Which one reacts first?
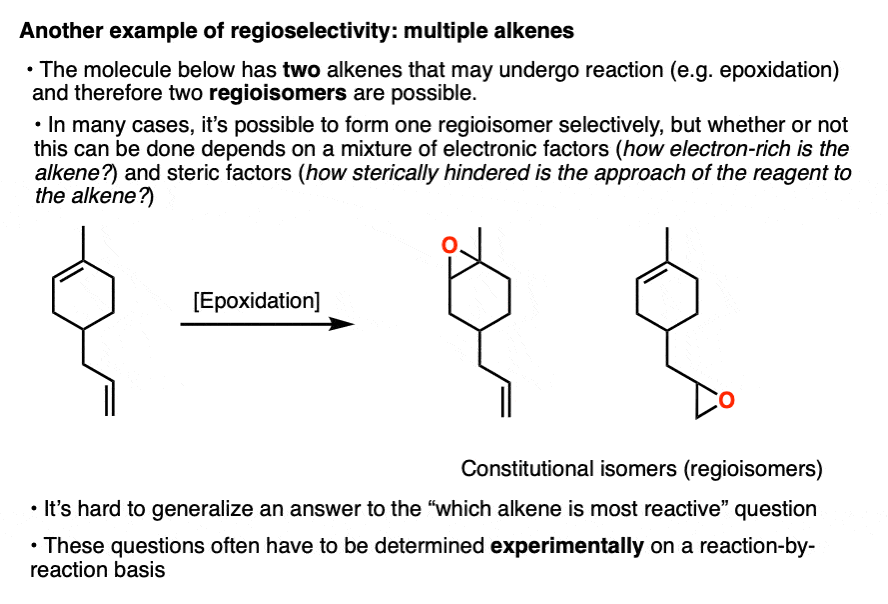
These types of questions usually have to be answered on a case-by-case basis since both steric and electronic factors can be at work. Organic chemistry is not math; we need to do experiments to really be sure!
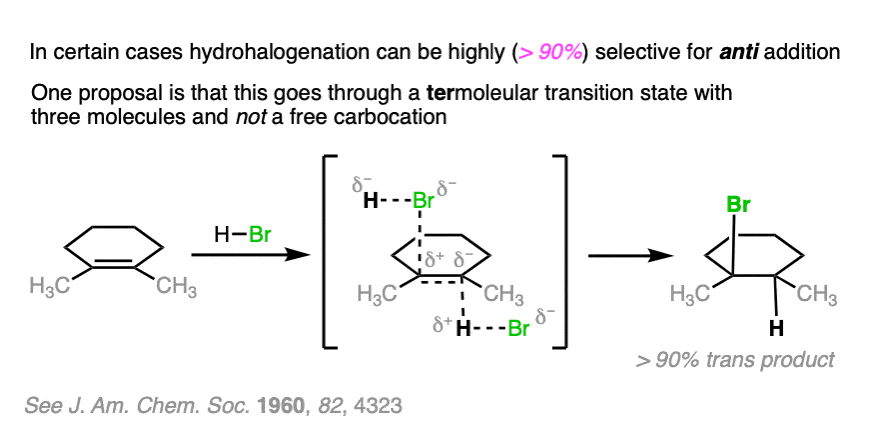
Note 4. . For the purposes of Org 1 / Org 2, we usually teach that alkene addition reactions with HX and the SN1 reaction are non-stereoselective and result in roughly equal mixtures of stereoisomers. In the real world, intimate ion pairing effects can result in both addition reactions of H-X and SN1 reactions to be at least somewhat stereoselective.
Note 5. The catalytic hydrogenation of alkenes with Pd/C and H2 is another example of a reaction that is generally stereoselective (for syn addition to alkenes) but in practice is not stereospecific. For more, see the footnotes in Palladium on Carbon for Hydrogenation of Alkenes.
Quiz Yourself!
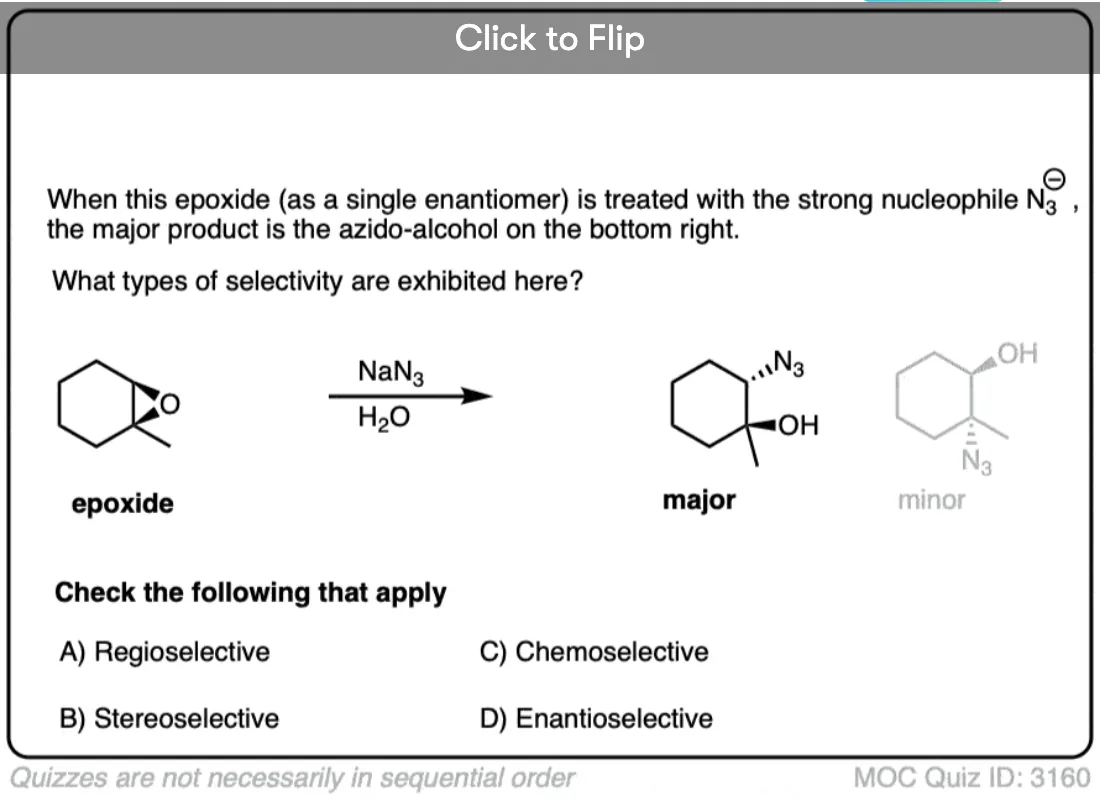
Become a MOC member to see the clickable quiz with answers on the back.
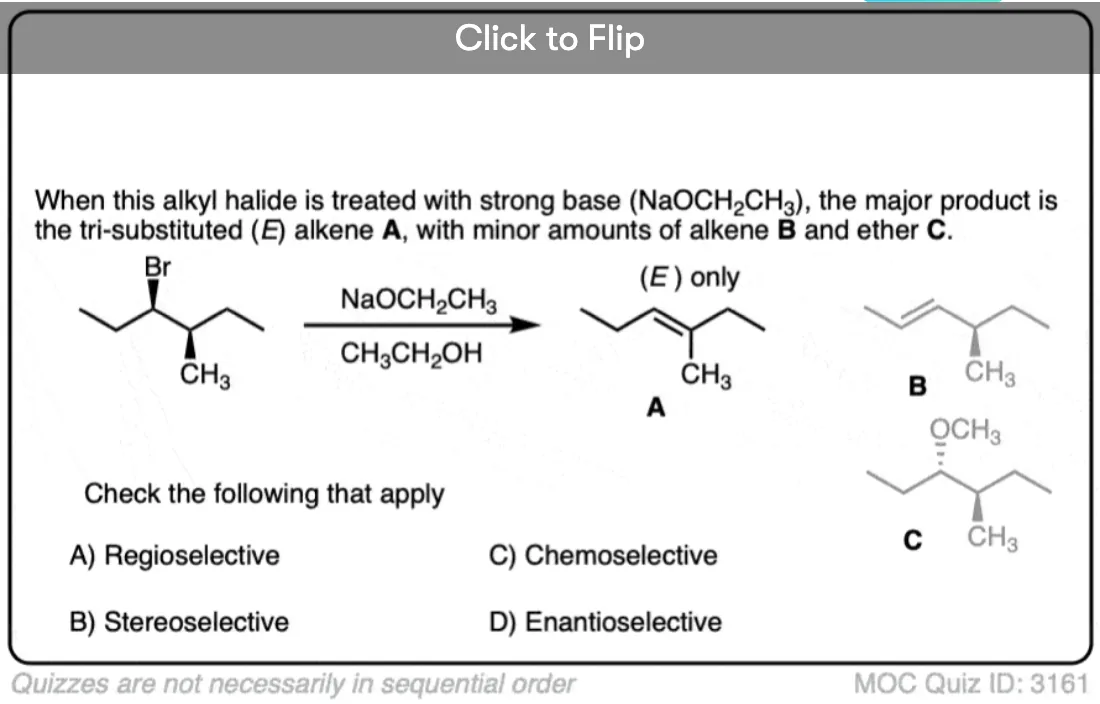
Become a MOC member to see the clickable quiz with answers on the back.
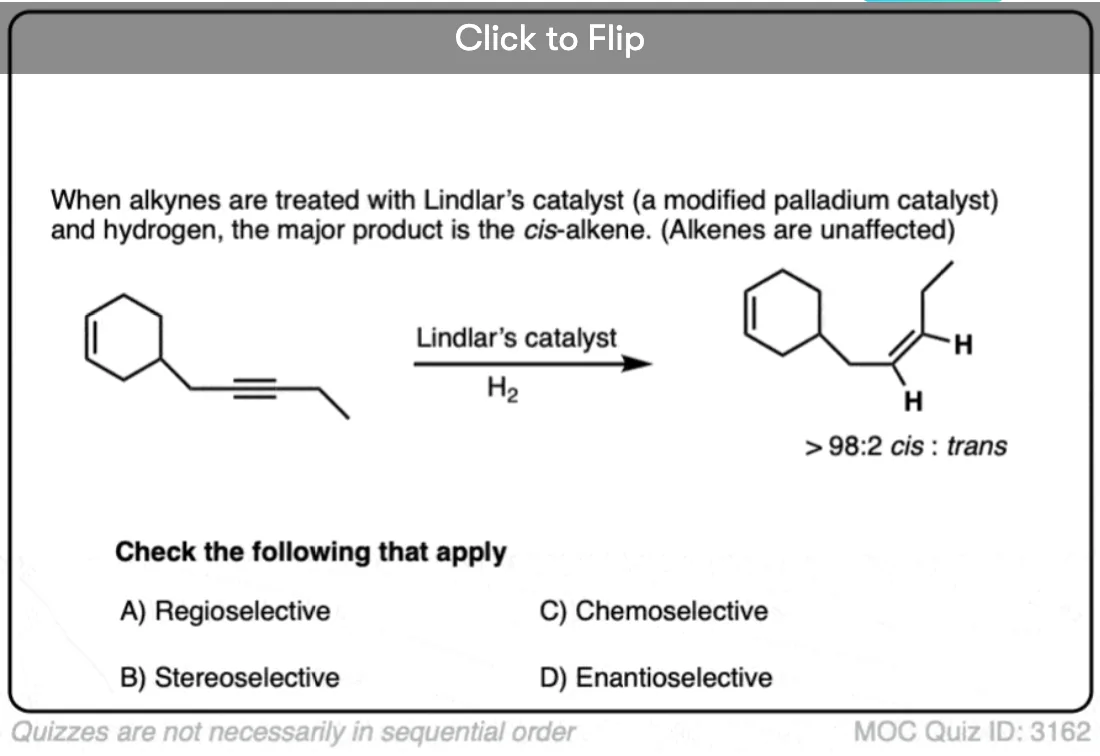
Become a MOC member to see the clickable quiz with answers on the back.
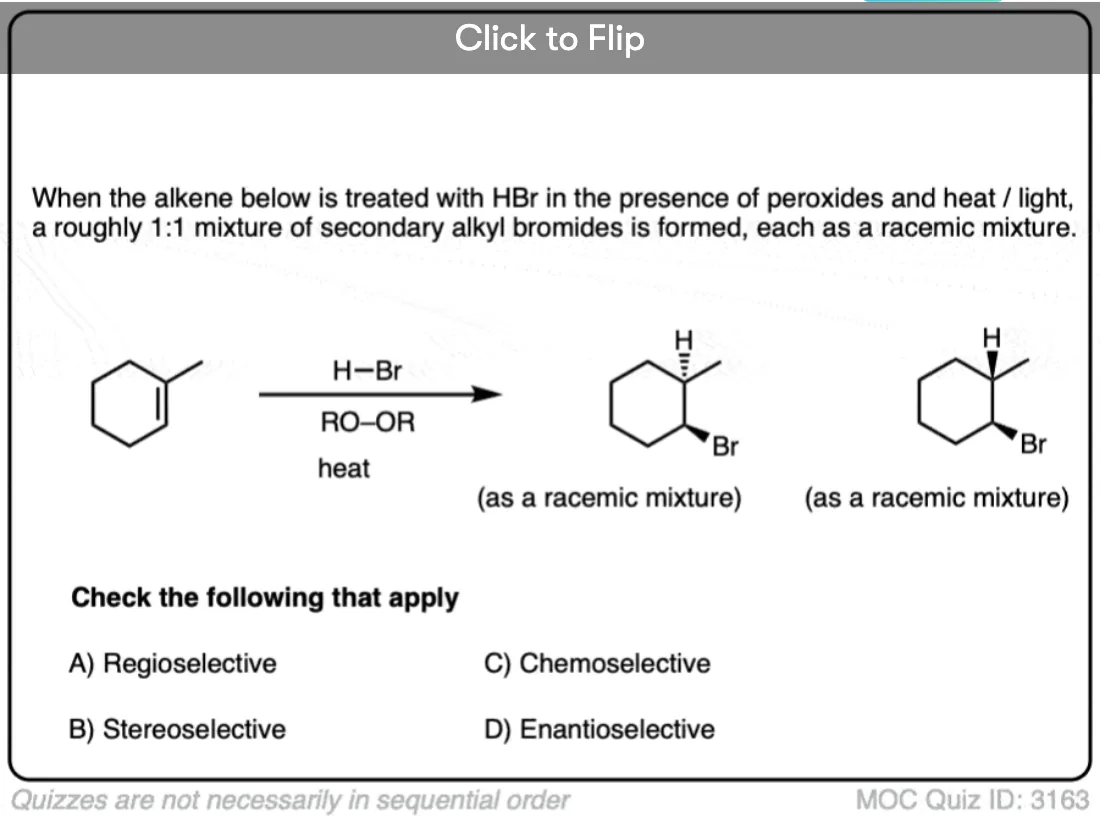
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading
- Notes from Prof. Andrew Myers’ course on the Sharpless Asymmetric Epoxidation.
- IUPAC – Stereospecificity
- IUPAC – Stereoselectivity
- The Nobel Prize in Chemistry 2022.
- Chemoselectivity: The Mother of Invention in Total Synthesis
Ryan A. Shenvi, Daniel P. O’Malley, and Phil S. Baran
Accounts of Chemical Research 2009 42 (4), 530-541
DOI: 10.1021/ar800182r
Some research stories from the Baran laboratory (Scripps) where chemoselective reactions have been achieved without the significant use of protecting groups. - Click Chemistry: Diverse Chemical Function from a Few Good Reactions
Hartmuth C. Kolb, M. G. Finn., K. Barry Sharpless
Angew. Chem. Int. Ed. 2001 40 (11), 2004-2021.
DOI: 10.1002/1521-3773(20010601)40:11%3C2004::AID-ANIE2004%3E3.0.CO;2-5
The article on selective reactions that led to the 2022 Nobel Prize.
You mentioned IUPAC frowns upon the use of the word “stereospecific” for dihydroxylation of alkenes with osmium tetroxide, but isn’t this reaction actually stereospecific even in IUPAC’s definition? When using Z and E alkenes, this reaction does give stereoisomeric products.
Can we call oxymercuration-demercuration(OMDM) as stereoselective?
The oxymercuration is stereoselective and stereospecific.
The demercuration process is not, since it involves a free radical with an indefinite lifetime that can potentially invert and give rise to mixtures of syn and anti addition.
Is this still stereospecific epoxidation (with eg mcpba) when we use terminal alkene that doesnt have stereoisomers?
H2c=ch-r
It would lead to a racemic mixture of enantiomers (assuming R is achiral).
thank you. I got it. Just now I checked this page and your reply. sorry for the delayed response.
can you explain this….a molecule has both primary and secondary amine, but only secondary amine or primary amine (specific) reacts with an electrophile. What do we call the reaction?
If you’re talking about selectivity for different amines (primary vs secondary) it would still be “regioselectivity” you’re talking about. If it were selectivity for an amine versus an alcohol (i.e. different functional groups) then that would be “chemoselectivity”.
It was very helpful ….and it would be glad …if u even emphasise on reactions that are both stereospecific and stereo selective…so that the topic becomes more clear .
A good suggestion. Thank you.
Why would you say hydroboration is regioselective? Is that based upon actual experiments and empirical data you’ve looked at? If the hydrogen has a partial negative charge with its bond with Boron, why would it add to the more highly substituted carbon instead?
It’s absolutely regioselective. For more information, see this post: https://www.masterorganicchemistry.com/2013/03/28/hydroboration-of-alkenes-the-mechanism/
Epoxidation is stereospecific and sharpless asymmetric epoxidation is stereoselective.explain
“A reaction is termed stereospecific if starting materials differing only in their configuration are converted into stereoisomeric products. According to this definition, a stereospecific process is necessarily stereoselective but not all stereoselective processes are stereospecific. ” That’s from IUPAC, here. https://goldbook.iupac.org/terms/view/S05994 .
That’s slightly different from the way the term is commonly used, as I believe it is in your case. Let me explain.
Epoxidation ALWAYS gives the “syn” epoxide, and NEVER the “trans”. It’s a consequence of the mechanism.
Take cis-2-butene and trans-2-butene and epoxidize them. cis-2-butene will give you cis-2-butene epoxide, and trans-2-butene will give you trans-2-butene epoxide, and there is no leakage between the two. That’s generally what’s meant by SPECIFIC.
In Sharpless epoxidation, you’re starting with an achiral alkene (actually allylic alcohol). After the Sharpless you might end up with a 99:1 mixture of enantiomers. Although 99:1 is very high, it’s not “specific”, it’s “selective”.
Like I said that’s the common usage, which is a little different from IUPAC’s sometimes.
Example of stereoselective reaction of alkynes??
Lindlar reduction to give cis-alkenes. Sodium metal reduction to give trans alkenes.
Is SN1 Stereoselective reaction or not?
In my opinion, an interesting addition to the explanation above is how IUPAC defines the terms (actually for me this is the only correct definition, even though it sometimes takes ages to really understand what it means).
Stereoselectivity is defined in http://goldbook.iupac.org/S05991.html.
Stereoselectivity has to do with the preferential formation of one stereoisomer over the other in a reaction.
Stereospecificity on the other hand (defined in http://goldbook.iupac.org/S05994.html) has to do with the fact that compounds that differ in their configuration (i.e. that are stereoisomers of each other) give a different reaction outcome in terms of stereochemistry of the products (cis- and trans-2-butene give different compounds upon reacting with bromine).
Using stereospecific in the sense of ‘with high stereoselectivity’ is actually discouraged by IUPAC. Here one could maybe prefer to use the terminology highly stereoselective (probably also ill-defined) or completely stereoselective depending on the case under investigation.
This is illustrated in the following example: the reduction of 2-butyne with H2 in the presence of a Lindlar catalyst is a completely stereoselective reaction: the cis-alkene is exclusively formed. This however is NOT a stereospecific reaction (at least not in the IUPAC sense of the word) as 2-butyne does not have a stereoisomer that could react differently.
Hi James, great article. I’d like to cite you in an assignment I’m doing for my Chemistry degree. How would i do that please?
Just cite the URL.
after observing the reaction how we can say that its a stereoselective or stereospecific reaction?
I wanted to know…
If the reactions that proceed through carbocation formation can be stereoselective…For example,The dehydration of alcohols thorugh E1 mechanism.Will this be anti elmimation similar to the case where the same reaction proceeds via E2.?
Very good and useful information
Hi, i have some confusion regarding stereospecificity. Isnt it different stereoisomer reactant gives different stereoisomeric products,then the reaction is stereospecific. example, (R)-reactant gives the configurationally inverted (S)-product, and (S)-reactant produces (R)-product. but why epoxidation of cis alkene produce only cis-product?
Hi James! I just had a quick question in regards to the example with 2.2.1 bicyclic compound example that you used. I actually thought that you would get predominantly the dashed product because of the “pointy part” potruding towards the top face of the molecule makes the top face more sterically hindered no?
Hi Frank – thanks for your question. The addition to the top face (“exo”) is favored – if you look at the bottom addition (“endo”) you’d see that instead of a one-carbon bridge getting in the way, there’s a two-carbon bridge getting in the way. For this reason attack is preferred on the same side as the 1 carbon bridge (“endo”). Interestingly though, if you put 2 methyl groups on the 1 carbon bridge the stereochem flips to endo.
http://pubs.acs.org/doi/abs/10.1021/ja00726a031
Hope this helps! James
Ahh…! Yes it does! Thanks for clearing that up for me. I think I wasn’t looking at it the right way, but I can see it now. :]
Thank you very much. This helped a lot.
This site is really helpful! Thank you very much :)
In the stereoselective reaction, in regards to the major product, what about that face of the 2.2.1 bicycle makes it the least hindered?
its good….bt i want to know reaqctions which are stereospecific and also stereoselective…
reaction which is both stereospecific and stereoselective both is the reaction between bromine and alkene……it is stereospecific because which stereoisomer we obtain depend upon which stereoisomeric alkene and stereoselective since from given alkene we obtain only one diastereomer or one pair of enantiomer.
All stereospecific reactions are stereoselective, since a sterespecific reaction gives a product ratio 100:0 and stereoselective reactions require any ratio other than 50:50.
A stereospecific reaction could be thaught as 100% setereoselective i.e, when the selectivity becomes 100% to a particular product of a stereoselective reaction. However a stereoselective reaction never can be called stereospecific.
All stereospecific reactions are stereoselective but all stereoselective reactions are not stereospecific.