Alkene Reactions
Hydrohalogenation of Alkenes and Markovnikov’s Rule
Last updated: May 28th, 2026 |
Hydrohalogenation of Alkenes and Markovnikov’s Rule
- When hydrohalic acids (HCl, HBr, HI) are added to alkenes, addition reactions can occur, resulting in formation of a C-H and C-halogen bond and breakage of a C-C pi bond.
- The reaction tends to occur such that the halogen ends up attached to the carbon of the alkene attached to the fewest hydrogen atoms, a phenomenon known as Markovnikov’s Rule.
- Addition of H-X to alkenes occurs through a protonation of the alkene to give a carbocation intermediate, followed by addition of the halide to the carbocation.
- A better reformulation of Markovnikov’s rule is therefore that addition of HX to alkenes will proceed through the most stable carbocation, which is generally the more substituted carbon of the alkene.
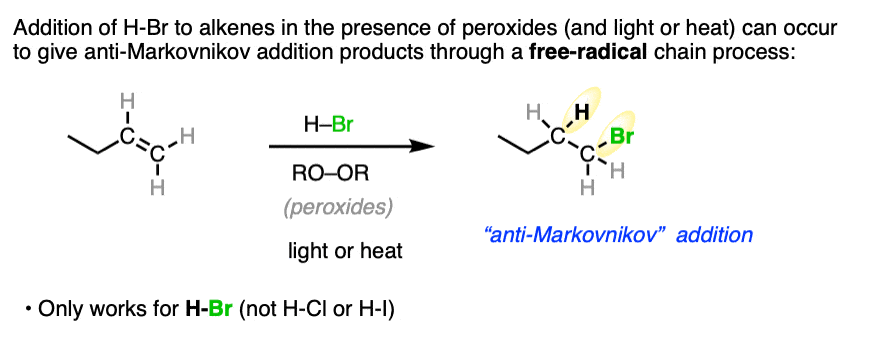
- (Note that H-Br with peroxides (RO-OR) operates through a different reaction mechanism – see Free Radical Addition of H-Br to Alkenes).
- Carbocation rearrangements (hydride or alkyl shifts) can occur if they will result in a more stable carbocation intermediate.
- H-X will also add to alkynes (See: Addition of HX to alkynes) and dienes (See: 1,2- and 1,4- Addition of HX To Dienes) but will not add to aromatic rings.
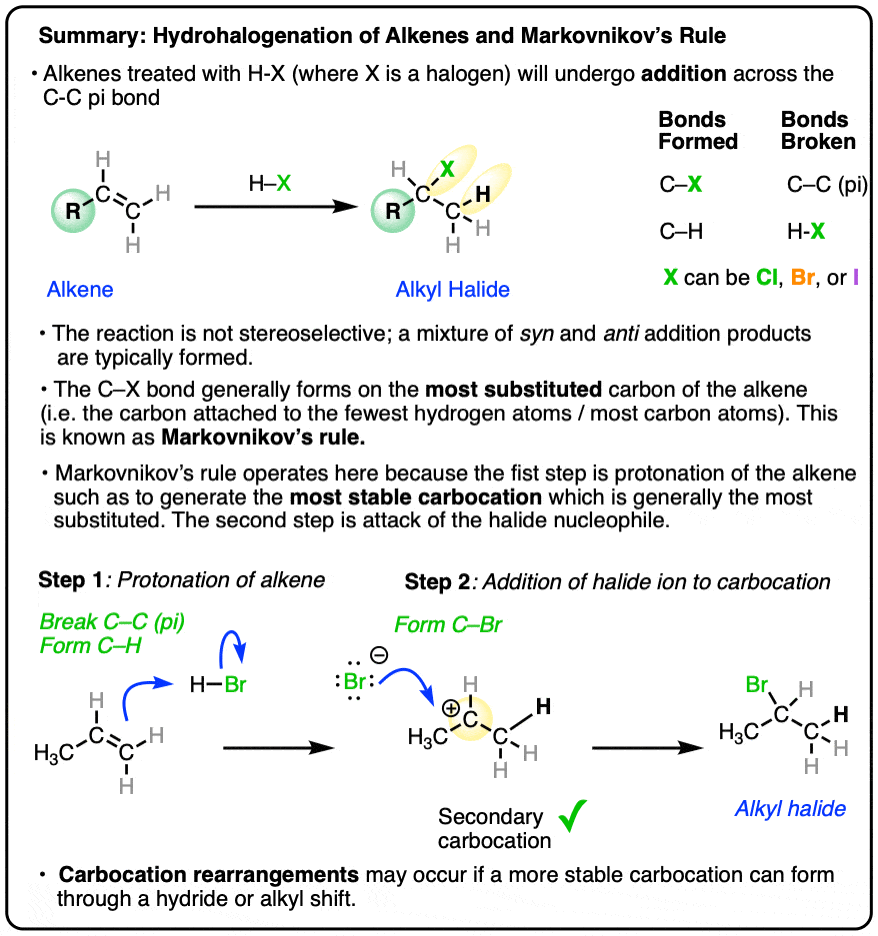
Table of Contents
-
- Reaction of Alkenes With Hydrohalic Acids (HCl, HBr, HI)
- Markovnikov’s Rule: The Halide Adds To The Most Substituted Carbon
- Stereoselectivity (or Lack Thereof) In Alkene Hydrohalogenation
- Hydrohalogenation of Alkenes: The Mechanism
- The Reaction Energy Diagram
- Carbocation Rearrangements – Hydride and Alkyl Shifts
- Alkynes, Dienes, Aromatic Rings, and the Cationic Cyclization of Alkenes
- Summary: Alkene Hydrohalogenation
- Notes
- Quiz Yourself!
- (Advanced) References and Further Reading
1. Reaction of Alkenes With Hydrohalic Acids (HCl, HBr, HI)
When alkenes are treated with hydrohalic acids (HCl, HBr, or HI) they form alkyl halides.
This is sometimes called hydrohalogenation of alkenes since it results in the addition of hydrogen and a halogen.
In these addition reactions:
- the C-C pi bond and the H-X bond breaks;
- a new C-H and C-X bond forms (where X is Cl, Br, or I).
If we add up the bond dissociation energies (BDE’s) of the bonds that form (C-H, about 98 kcal/mol, and C-Cl, about 81 kcal/mol) and subtract their sum from those of the bonds that break (C-C pi, about 60 kcal/mol, and H-Cl, about 103 kcal/mol) the reaction is exothermic by about 18 kcal/mol for the addition of HCl. [Link to a useful table]
Acid is required for these reactions to occur; no addition of halide to alkenes will happen in the absence of strong acid. (In other words, HCl = ✅, but NaCl = ❌).
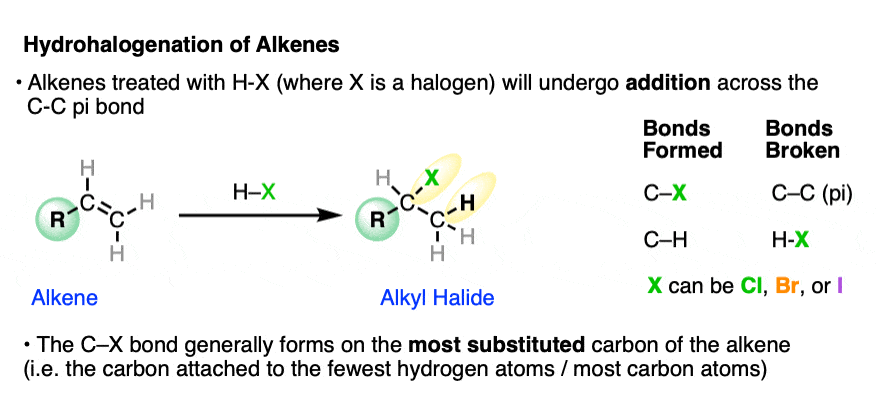
The new C-halogen bond tends to form on the most substituted carbon of the alkene (i.e. the one with the fewest carbons), and the new C-H bond tends to form on the carbon of the alkene containing the most hydrogens.
2. Markovnikov’s Rule: Regioselectivity of HX Addition to Alkenes
The reactions of alkenes with has been known for over 150 years.
When an alkene is not symmetrical, the reaction has the potential to form two different constitutional isomers (“regioisomers”). However, in many cases, one constitutional isomer is formed in a much higher proportion than the other. In other words, the reaction is regioselective.
In an early study of this reaction, Russian chemist Victor Markovnikov published the observation that the halogen tended to add to the carbon of the alkene which was bonded to the least number of hydrogens.
This has come to be known as “Markovnikov’s Rule“.
Markovnikov’s rule is what we would call an “empirical” rule. It was based purely on observed results, without any real underlying understanding of the mechanism of the reaction.
In later years people had a hard time replicating Markovnikov’s results with HBr, often finding that the least substituted regioisomer was formed instead! Later on, in the 1930s the cause of this aberrant regioselectivity was found to be the presence of peroxides in solvent, which led to a free-radical process for addition of H-Br to alkenes, which does not occur with H-I or H-Cl. See article – Free-Radical addition of H-Br to Alkenes. [Note 1]
Subsequently we tend to say, for better or worse, that addition reactions to alkenes that follow this pattern (such as their reaction with H3O+ ) are “Markovnikov-selective“, whereas reactions that follow the opposite pattern are “anti-Markovnikov selective” (such as hydroboration of alkenes).
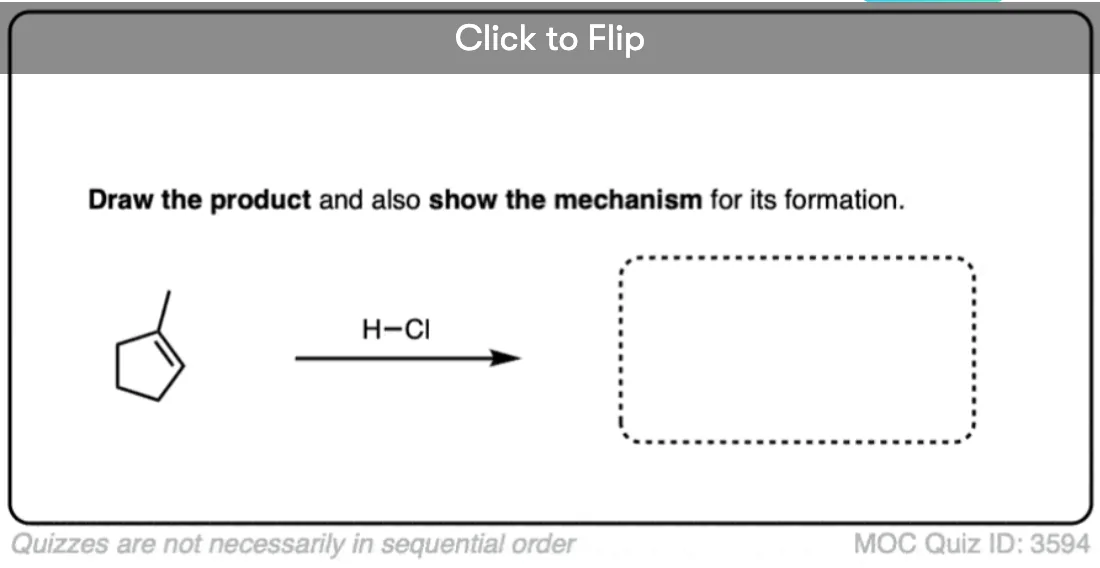
See if you can apply the pattern in the reaction of the alkene below (1-methylcyclopentene) with H-Cl.
 Click to Flip
Click to Flip

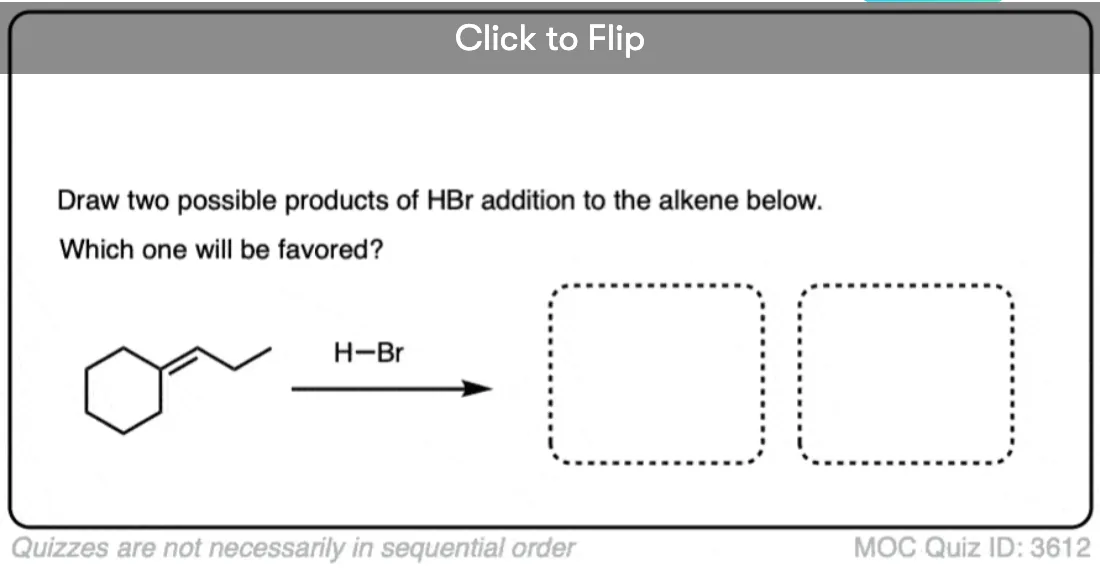
Another example is below. Draw the major product(s) and determine how they will be related:
 Click to Flip
Click to Flip

Can you work backwards to come up with the best starting material for this reaction?
 Click to Flip
Click to Flip

3. Stereoselectivity (Or Lack Thereof) In Addition of HX To Alkenes
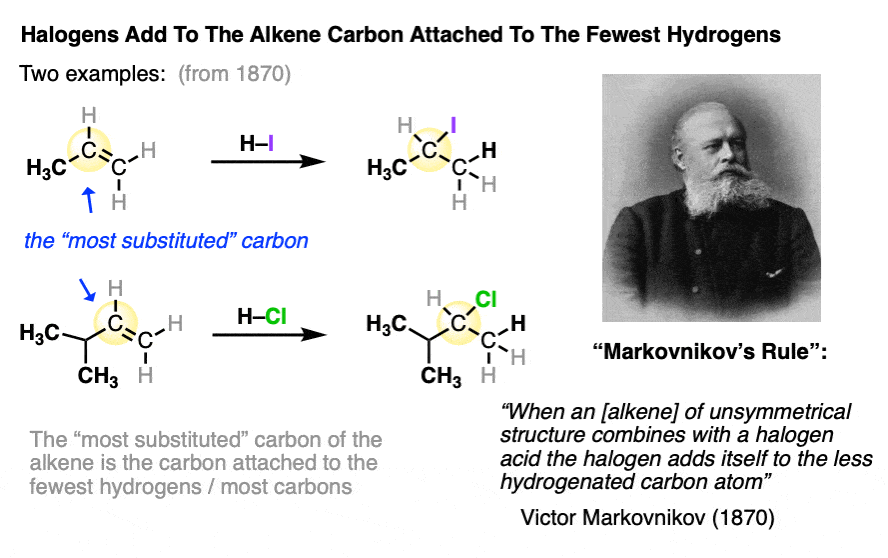
When addition reactions occur across an alkene pi-bond, the (sp2-hybridized) trigonal planar carbons of the alkene are converted into (sp3-hybridized) tetrahedral carbons, and with this comes the potential for formation of stereoisomers. (See article: Types of Isomers)
- When the two new bonds to carbon are formed on the same face of the alkene, the addition is said to be “syn“
- When the two new bonds are formed on opposite faces of the alkene, the addition is said to be “anti“.
Addition reactions that give primarily syn or anti products are said to be stereoselective, and we will meet many examples in subsequent articles in this chapter (e.g. halogenation is stereoselective for anti products, and hydroboration is stereoselective for syn products).
Addition of H-X to alkenes gives a mixture of syn addition products and anti addition products [Note 2].
In other words, the reaction is not particularly stereoselective.
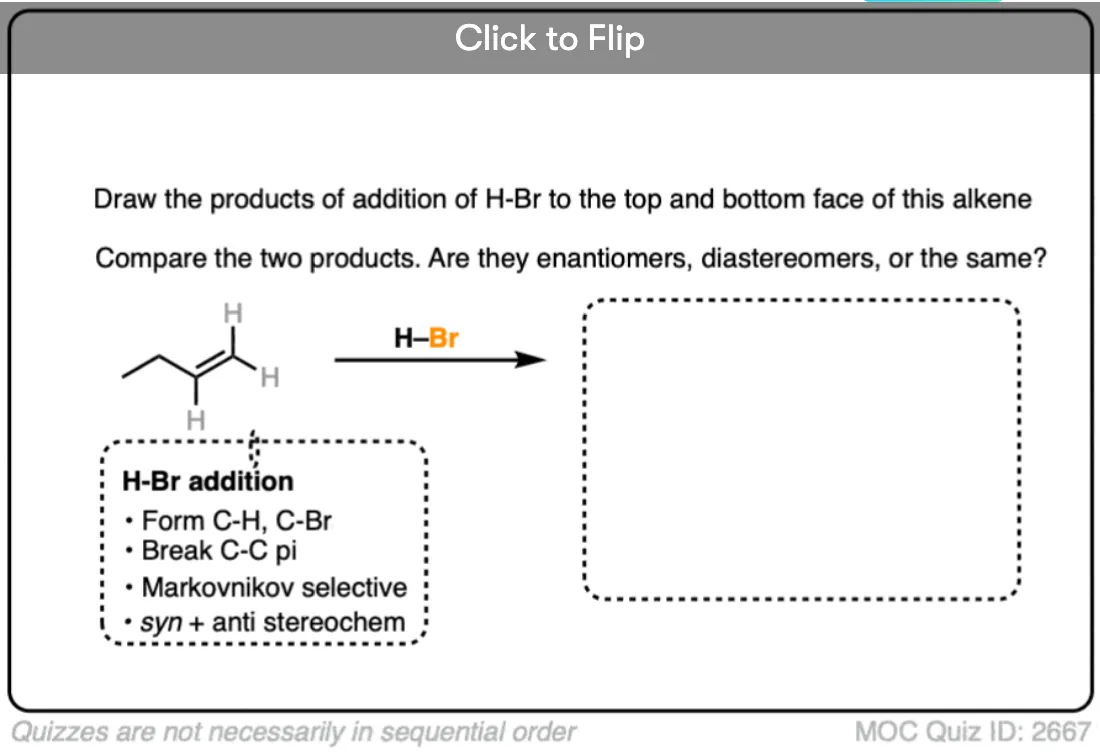
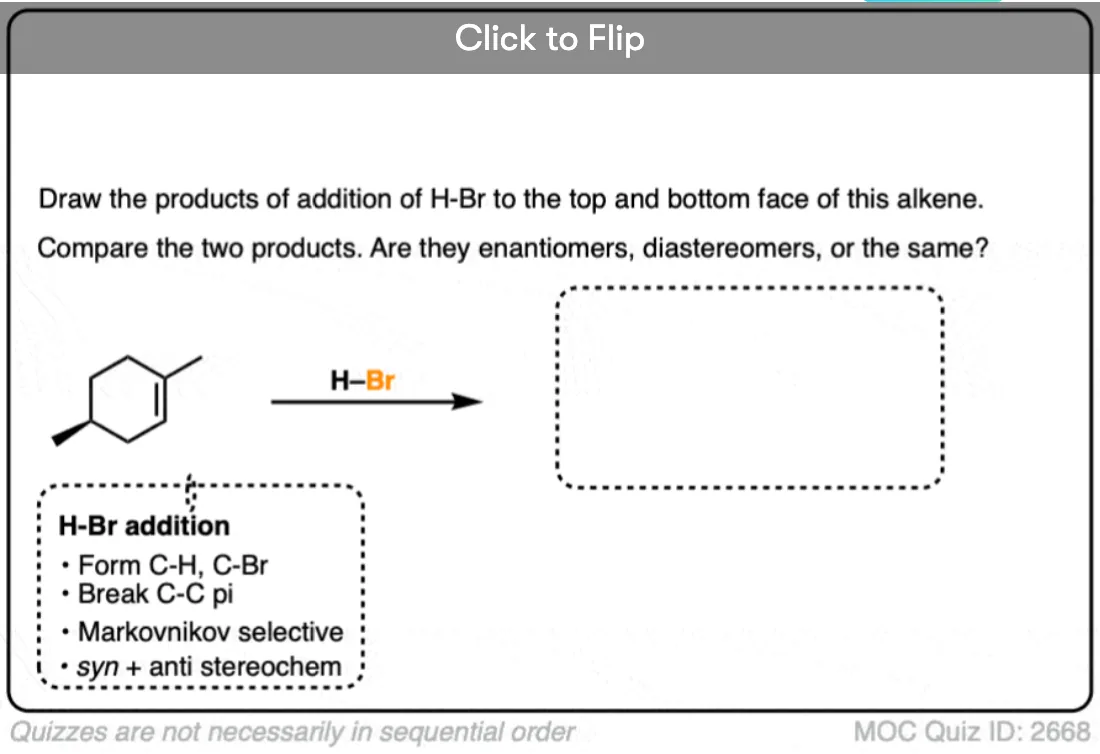
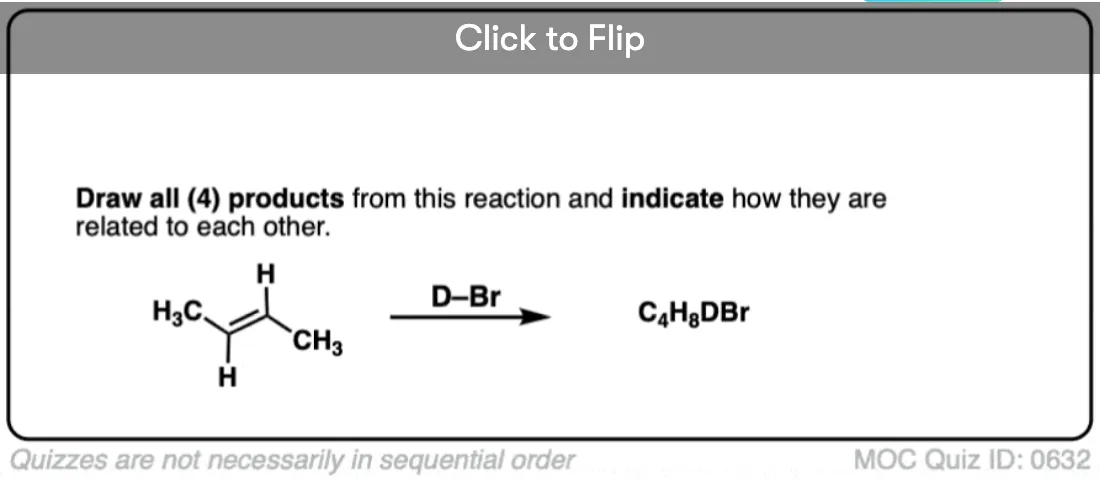
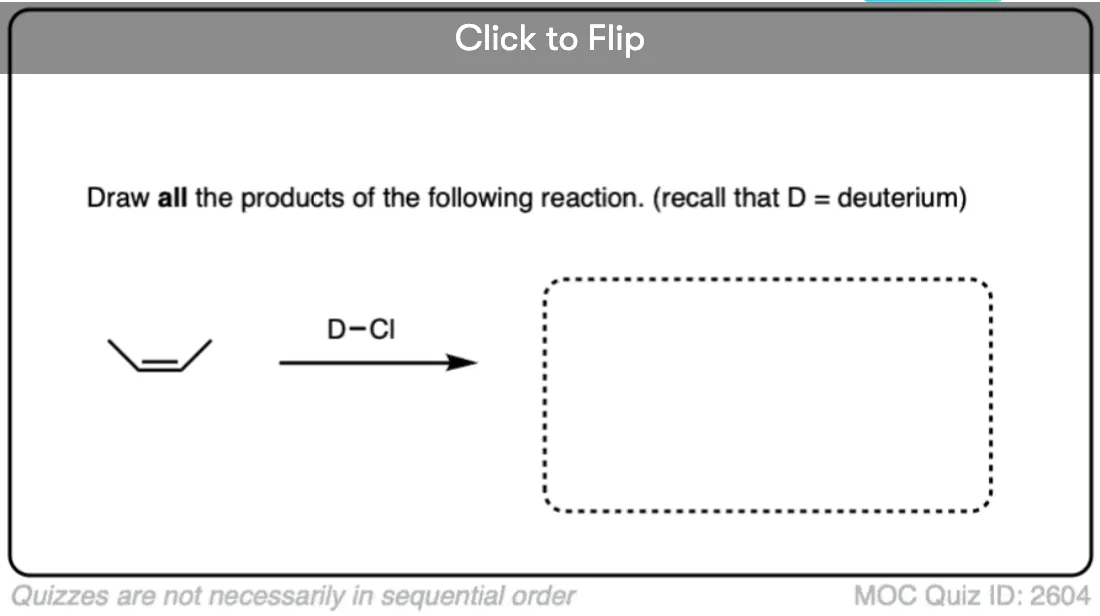
This lack of stereoselectivity leads to mixtures of syn and anti addition products, in some cases each as mixtures of enantiomers. See if you can draw the products of the reaction below:
 Click to Flip
Click to Flip

4. Hydrohalogenation of Alkenes: The Mechanism
Markovnikov was one of the more prominent chemists of his era, but back in 1870 he didn’t know why the reaction tended to give the more substituted product. The first to really figure it out was Lucas in 1924 [Ref], based on observations that alkyl groups are more “electron releasing”.
After many decades it was finally proposed that reaction goes through an intermediate carbocation. (The existence of carbocations was a fairly controversial subject until the 1930’s).
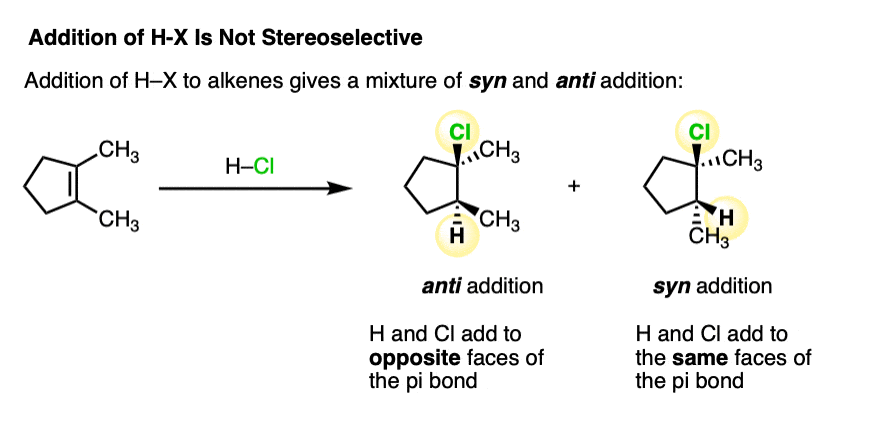
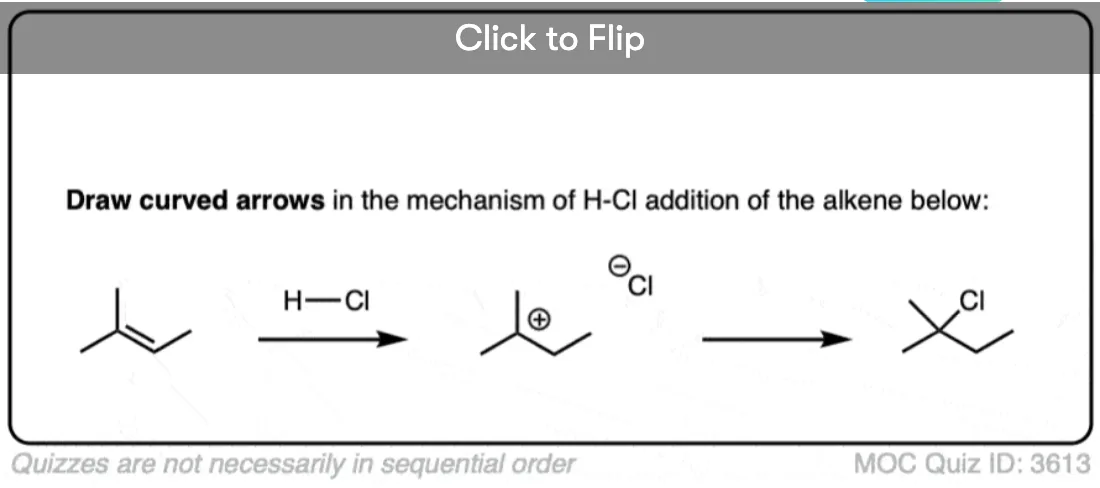
In the first step, the alkene (a nucleophile) is protonated by strong acid, resulting in a new carbocation. [Note 3 ]
Depending on where protonation occurs, two different carbocations may be formed. Being electron-poor species, carbocations are stabilized by adjacent electron-donating groups as well as by delocalization through resonance. (See article – 3 Factors Which Stabilize Carbocations).
The transition state leading to the most stable carbocation will be lower in energy, which tends to be the most substituted carbocation.
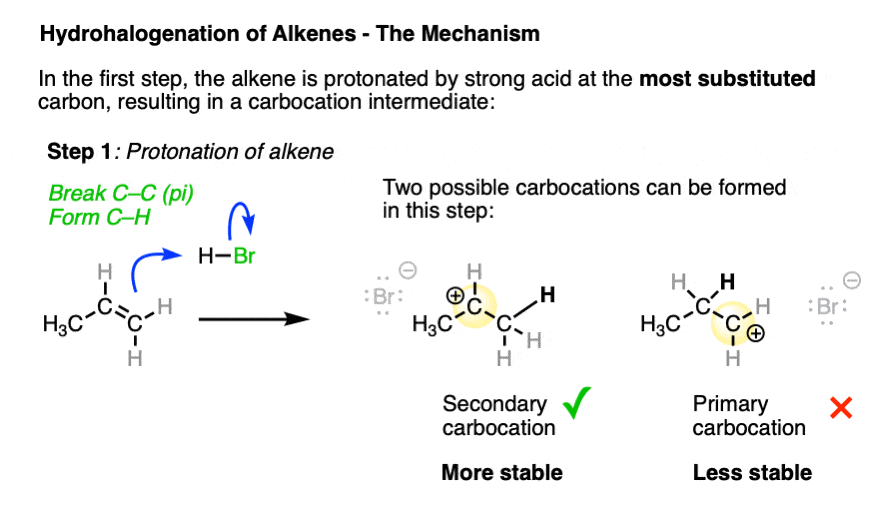
The arrow pushing for alkenes reacting with acids like H-X can be a little ambiguous – see Note 4.
So it is ultimately this carbocation intermediate which is the underlying reason for why Markovnikov’s rule is observed.
Carbocations have an empty p-orbital and readily accept a pair of electrons from whatever Lewis bases happen to be present in solution.
In the second step, the best nucleophile present (which tends to be the halide ion) then attacks the carbocation, forming the alkyl halide.
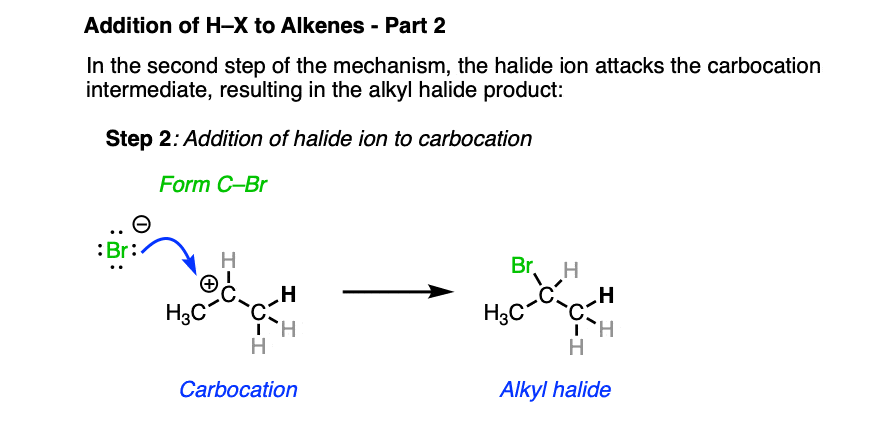
To reiterate: Markovnikov went to his grave not knowing this mechanism. Heck, many chemists of his time didn’t even accept that carbon was tetrahedral! So if you didn’t figure this mechanism out immediately, that’s to be expected and certainly nothing to feel bad about. The body of knowledge that is chemistry is built up of thousands of little experiments that eventually grew into the framework we have today.
5. Reaction Energy Diagram For Alkene Hydrohalogenation
Sometimes it can be helpful to trace out the energy profile of a reaction as it progresses from starting material to products.
In these diagrams, peaks (local maxima) are transition states and valleys (local minima) are intermediates.
A simple hydrohalogenation reaction has two steps – each of which has a transition state – and a single carbocation intermediate.
- In the first step, our intrepid alkene is protonated by strong acid, resulting in the high-energy transition state TS 1. This step has the highest activation energy (the barrier between reactants and transition state) making it the rate-determining step for this reaction.
- From the transition state (TS1) energy maximum, the reaction proceeds to the carbocation intermediate.
- The second step involves attack of the halide nucleophile on the carbocation, which proceeds through transition state 2 (TS2). Note that the activation energy for this step is considerably less than for protonation of the alkene – in other words, it will be the fast step.
- The reaction then proceeds through TS2 to give the final product, the alkyl halide.
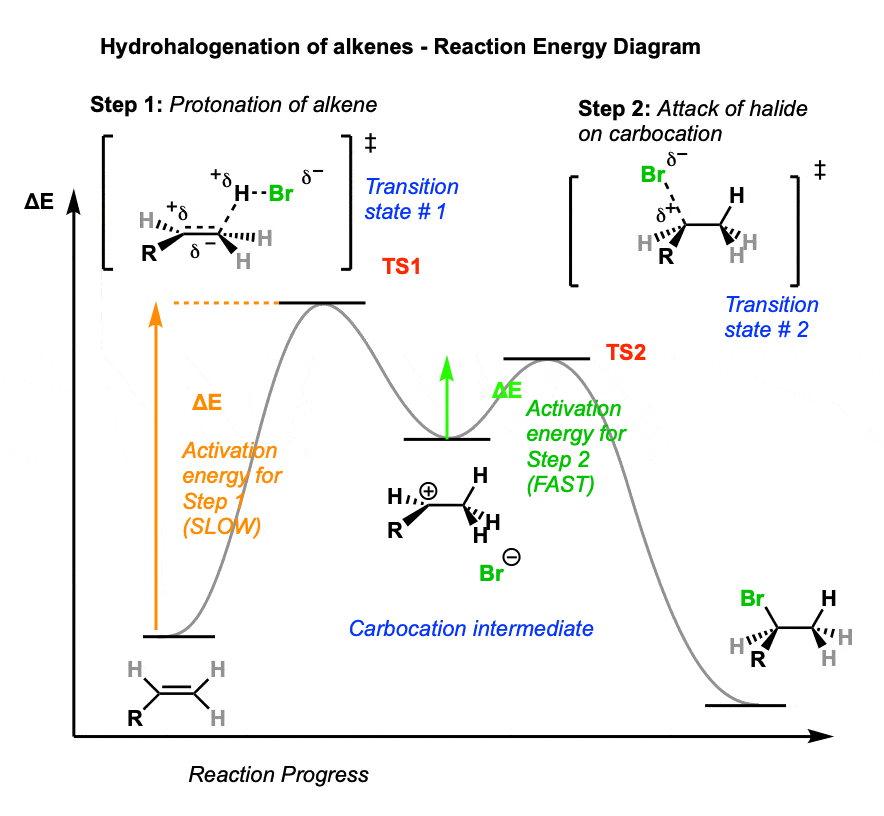
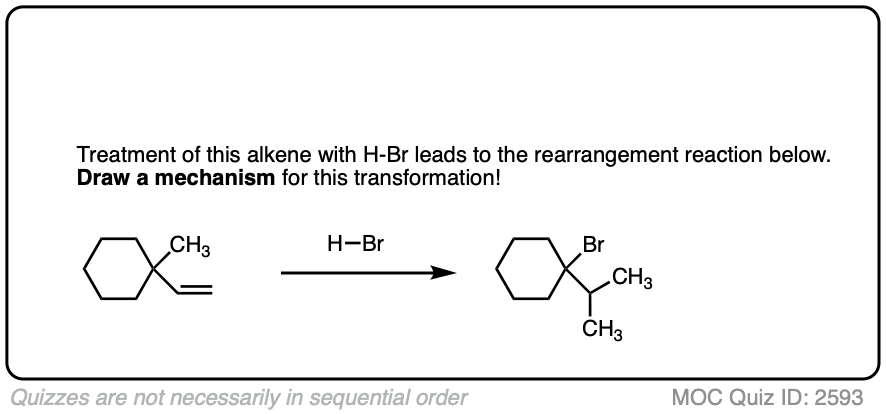
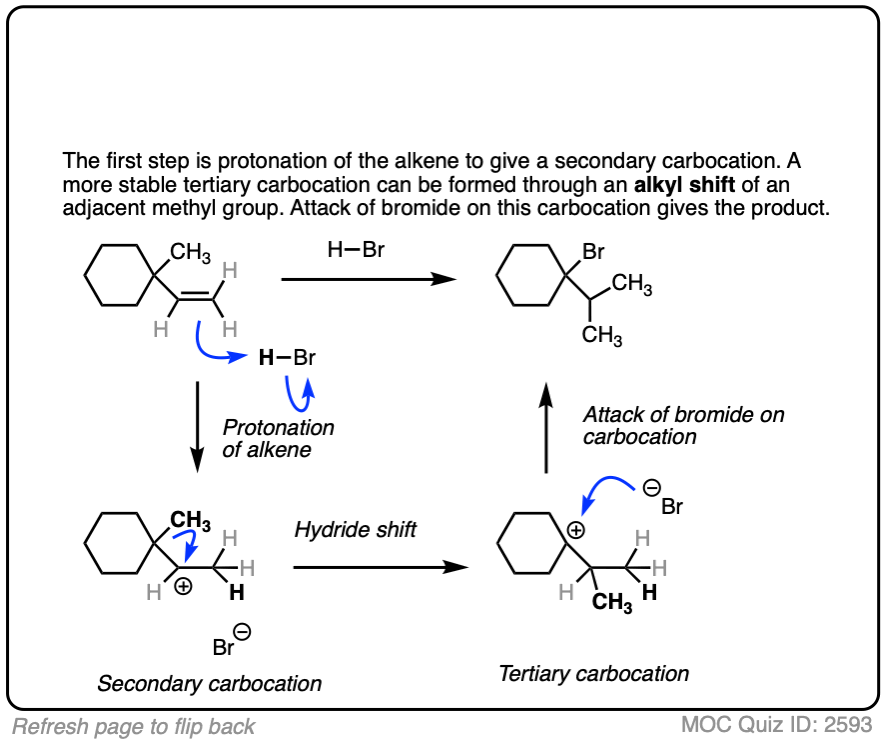
6. Carbocation Rearrangements – Hydride and Alkyl Shifts
One key to the proposal of a carbocation intermediate was the observation that some hydrohalogenation reactions give products of carbocation rearrangements in addition to the expected addition product.
For example when 4-methyl-1-butene was treated with HCl and allowed to sit at room temperature for an extended period, the product mixture was found to contain about half the expected Markovnikov addition product in addition to a new tertiary alkyl halide. [Ref]
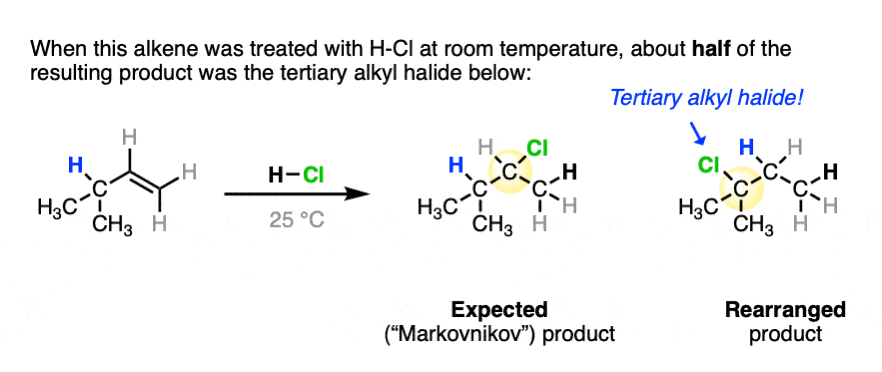
In this reaction the C-H bond that was originally part of the isopropyl group migrates to the secondary alkyl carbon. (See article – Rearrangements in Alkene Addition Reactions)
See if you can draw a reasonable mechanism!
 Click to Flip
Click to Flip

These rearrangements can occur when a more stable carbocation can result from migration of a hydride or alkyl group. When a quaternary carbon is adjacent to a secondary carbocation, alkyl groups can migrate.
 Click to Flip
Click to Flip
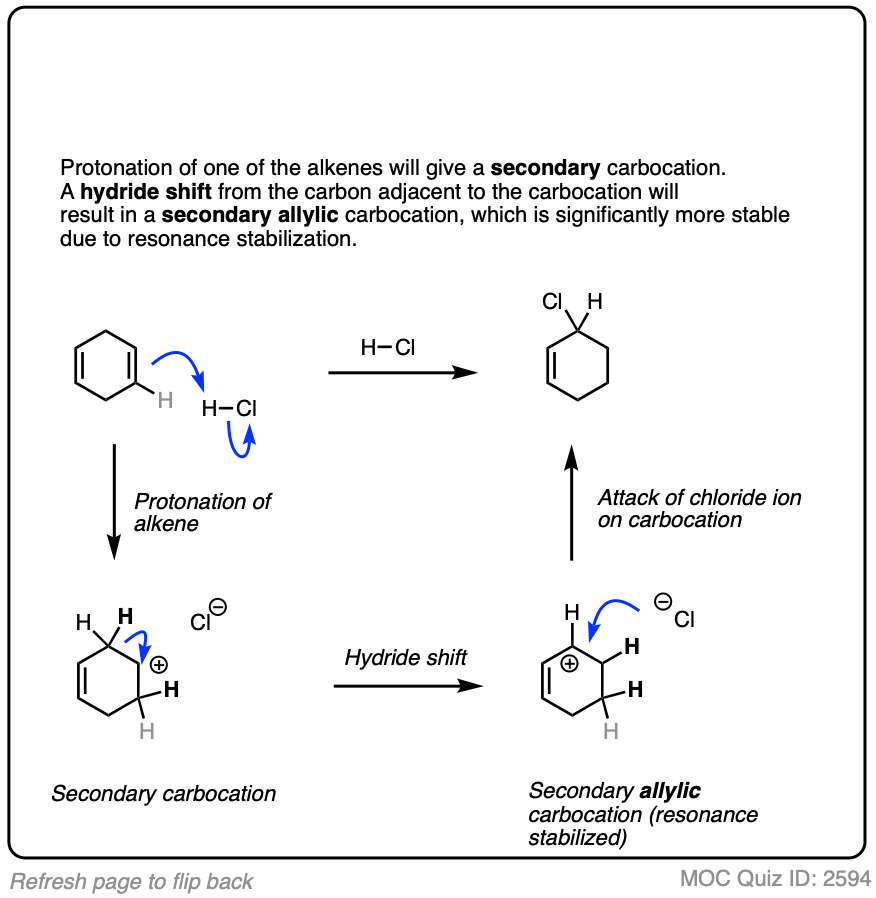
Occasionally migrations between equivalently substituted groups can be favored, as in the example below.
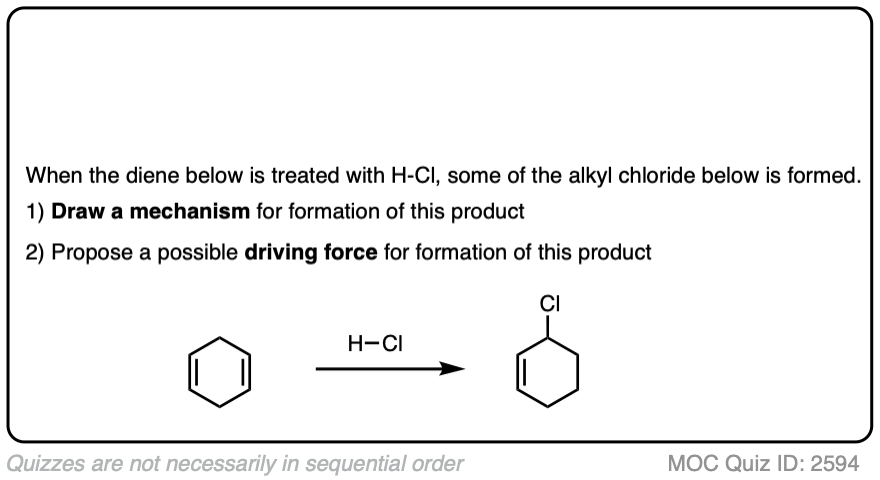 Click to Flip
Click to Flip
7. Other Applications of Hydrohalogenation
- Alkynes will react with HX to give vinyl halides. A second equivalent of HX will give geminal dihalides. (For more, see Alkyne Hydrohalogenation)
- Dienes such as 1,3-butadiene will react with HX to give various products. See this article in the chapter on conjugated systems for more. (See article: 1,2- and 1,4-Addition of HX To Dienes)
- Aromatic rings such as benzene will not undergo addition reactions with HX. Aromatic rings tend to react through substitution. More in the chapter on aromatic rings. (See article: Introduction to Aromaticity)
Carbocations are reactive intermediates and will readily combine with even poor Lewis bases.
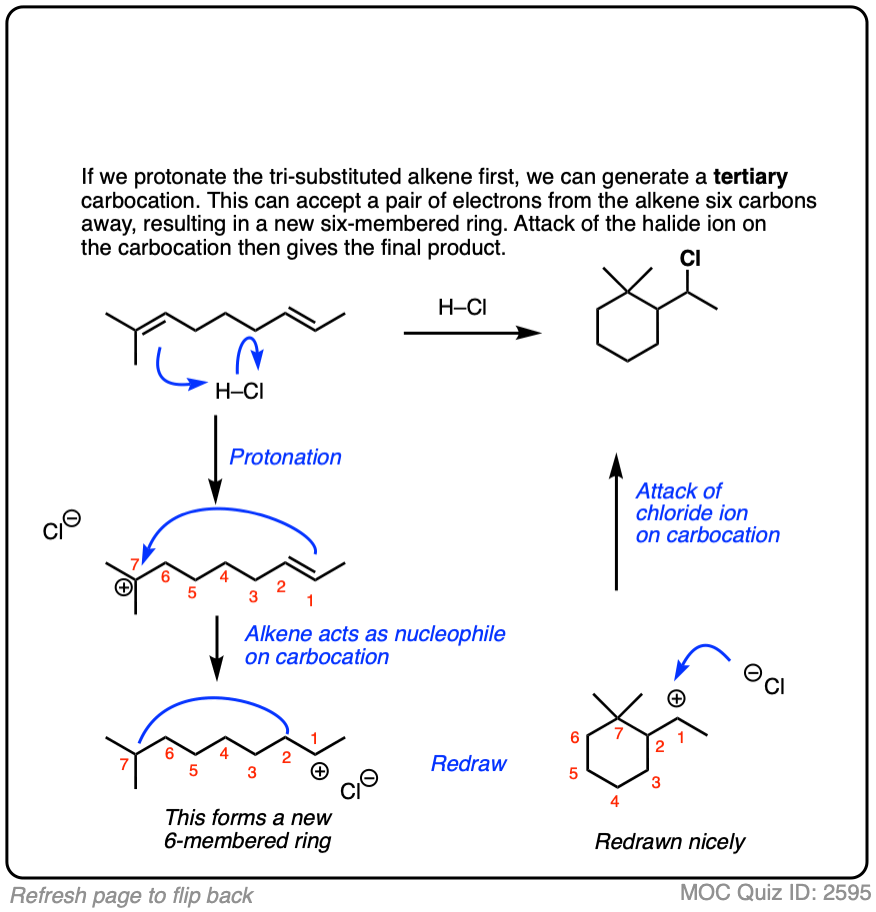
Sometimes those Lewis bases include other alkenes on the same molecule. This can result in cyclic molecules.
For example, consider the reaction below:
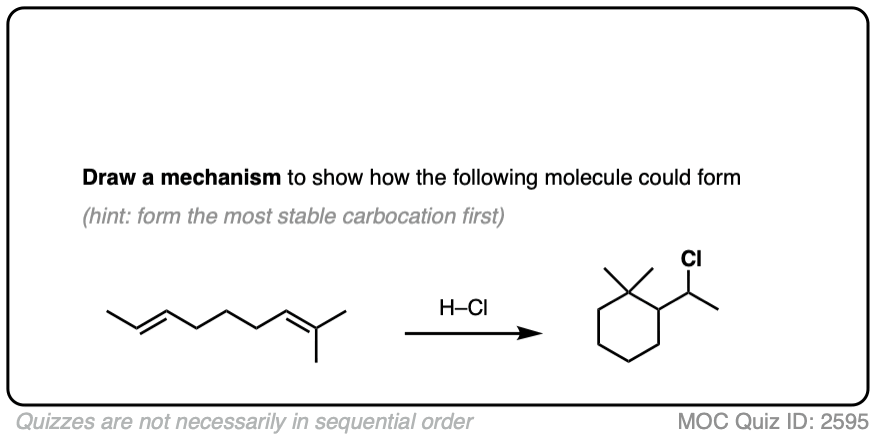 Click to Flip
Click to Flip
There are whole classes of molecules that are synthesized in nature via attack of alkenes on various carbocations. One of the most prominent classes is terpenes. I don’t want to get into it in this article, but if you are looking for a good time, I strongly suggest looking at how the steroid skeleton of lanosterol is built up from the cyclization of squalene. [Note 5]
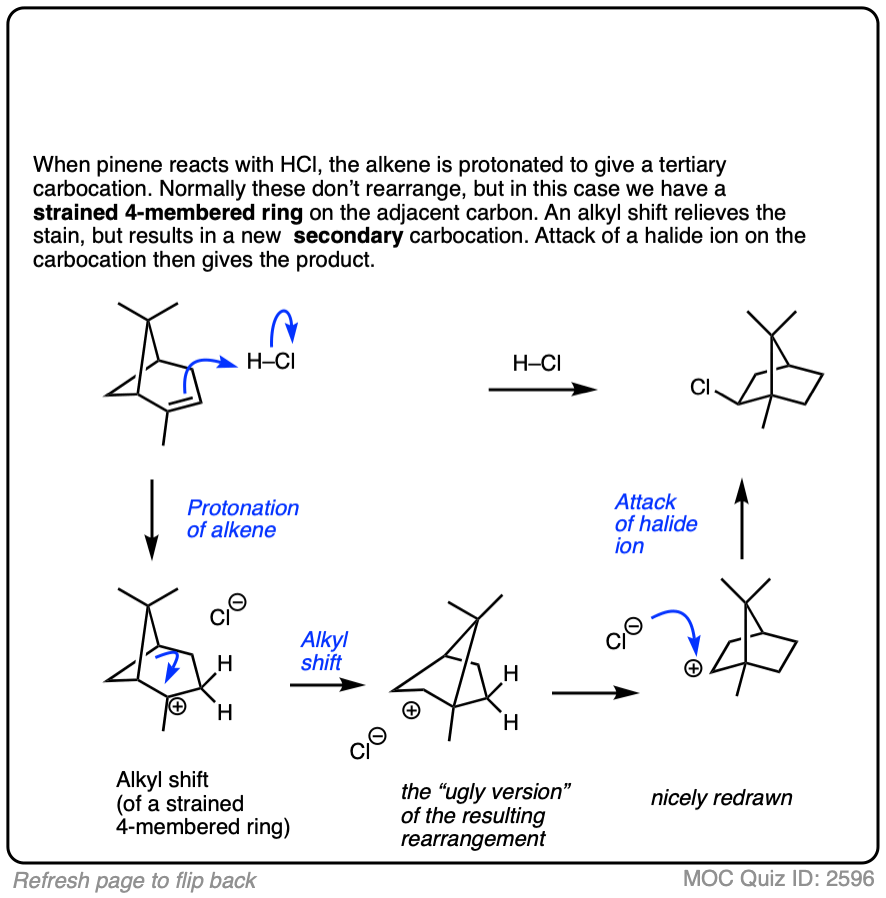
This isn’t the cyclization of an alkene, but it’s another example of how rearrangements can occur in nature. In this example we start with alpha-pinene, one of the main ingredients in pine oil.
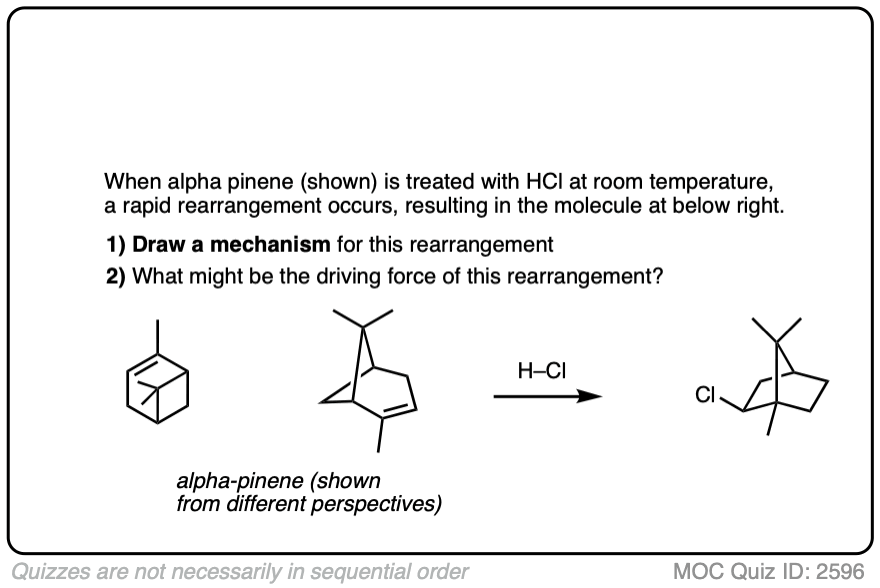 Click to Flip
Click to Flip
8. Summary
- HCl, HBr, and HI will all add to alkenes. The alkene acts as a nucleophile, forming a bond with the electrophilic proton of the acid.
- The reaction always occurs so as to form the most stable carbocation. This is responsible for the observations that led Markovnikov to postulate his rule in the first place.
- Be alert for the possibility of carbocation rearrangements when a more stable carbocation can be formed. This can sometimes even occur between carbons with similar substitution if it results in a resonance-stabilized carbocation.
- The reaction of HX with alkenes is essentially the same as the reaction of H3O+ with alkenes. They both follow what I call the Carbocation Pathway, one of the three key “buckets” of alkene reaction mechanisms that you might find helpful to remember. (See – Alkene Addition Reactions – The “Carbocation” Pathway)
Notes
Note 1. When peroxides (often written RO-OR) are present a very different reaction pathway occurs, where free-radical intermediates are involved. These reactions are anti-Markovnikov selective. For more on this reaction, see Free-Radical Addition of HBr To Alkenes.
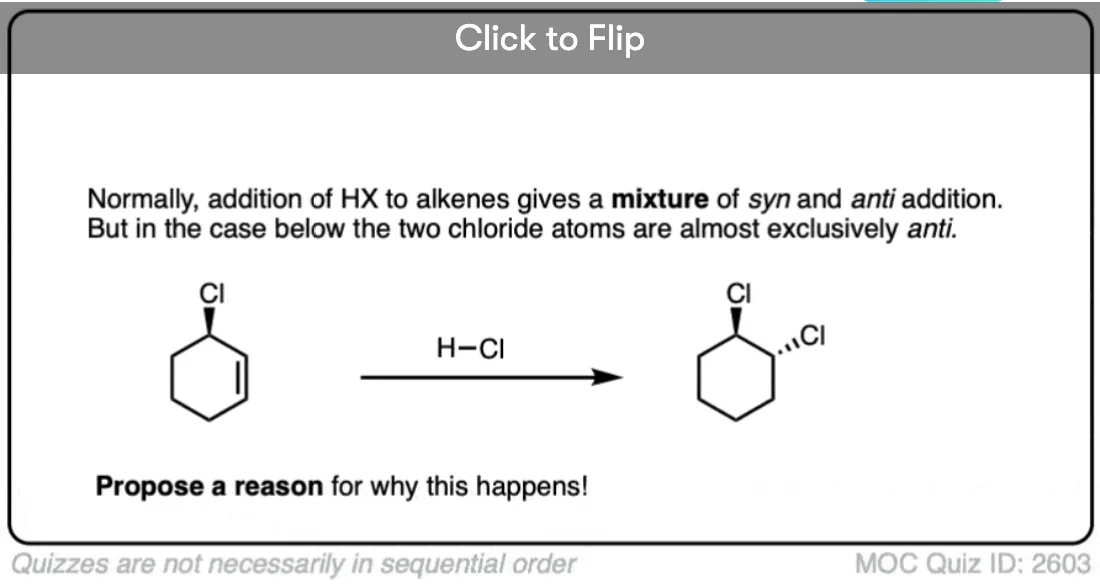
Note 2. Telling a small fib here, as this reaction does not always lack stereoselectivity. One particularly interesting set of results was in the addition of HBr to 1,2-dimethylcyclohexene in (the very non-polar solvent) pentane. It was found that addition occurred rapidly to give almost exclusively the trans product; letting it sit around for awhile led to some equilibration between stereoisomers.
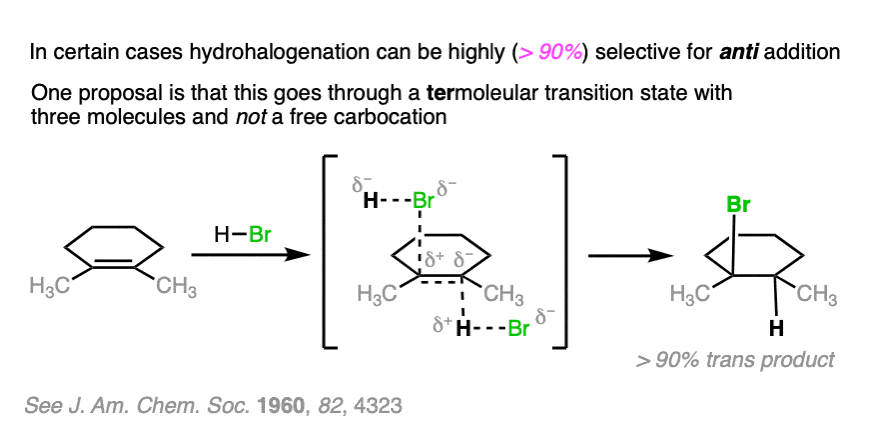
The authors propose that the reaction proceeds through a termolecular transition state (i.e. involving three molecules) with the alkene sandwiched between two molecules of HBr.
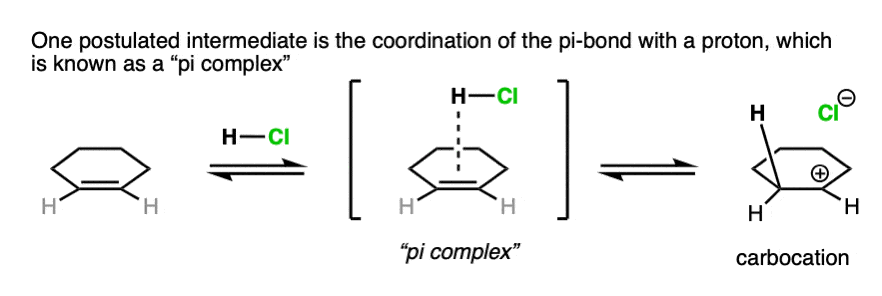
Note 3. For our purposes, drawing the immediate formation of a carbocation is fine. Some studies look for the possibility of an even earlier intermediate called a “pi complex”, where H+ is coordinated to the pi-bond. As the bond between the pi-bond to the proton becomes stronger, the pi-complex then evolves into the carbocation intermediate.
Note 4. Alkenes present a little bit of a dilemma in using curved arrows to describe the movement of electron pairs, since the double bond is not polarized and there is no obvious nucleophilic or electrophilic end. The convention in drawing arrows is that the carbon involved in bond formation is the one closest to the electrophile.
For example here is an example of arrow-pushing that shows the right-hand carbon of the double bond forming a new bond to H:
![]()
And here is an example of curved arrows showing formation of a new bond to the left side of the alkene:
![]()
To solve this dilemma, various new conventions have been devised such as the bouncy arrow formalism and drawing a dashed line to indicate the bond being formed, although these have not exactly taken the world by storm.
Note 5. If you are looking for a good time, this review outlines the whole rich and fascinating history of “cationic” alkene cyclizations, with a particular focus on how steroids and other related compounds are synthesized in nature.
Quiz Yourself!
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading
Ingold’s “Structure and Mechanism in Organic Chemistry” is a valuable guide to the early literature on this topic.
- The Logic Behind Markovnikov’s Rule: Was It an Inspired Guess? …No!
D. E. Lewis, Angew. Chem. Int. Ed. 2021, 60, 4412.
DOI: 10.1002/anie.202008228
Fun, accessible historical essay examining Markovnikov’s studies in the 1860’s-1870’s. - I. Ueber die Abhängigkeit der verschiedenen Vertretbarkeit des Radicalwasserstoffs in den isomeren Buttersäuren.
Markownikoff, W. (1870)
Justus Liebigs Ann. Chem., 153: 228-259.
DOI: 10.1002/jlac.18701530204
The original Markovnikov paper. - ELECTRON DISPLACEMENT IN CARBON COMPOUNDS I. ELECTRON DISPLACEMENT VERSUS ALTERNATE POLARITY IN ALIPHATIC COMPOUNDS
Howard J. Lucas and Archibald Y. Jameson
Journal of the American Chemical Society 1924 46 (11), 2475-2482
DOI: 10.1021/ja01676a018
If not the earliest explanation of Markovnikov’s rule, certainly one of them. - Secondary Isoamyl Chloride, 3-Chloro-2-methylbutane
Frank C. Whitmore and Franklin Johnston
Journal of the American Chemical Society 1933 55 (12), 5020-5022
DOI: 10.1021/ja01339a053
One of the first clearly written out explanations of a carbocation rearrangement in addition of HX to alkenes. A subsequent paper ( JACS 1950 1511) goes into more detail. - The Stereochemistry of the Addition of Hydrogen Bromide to 1,2-Dimethylcyclohexene
George S. Hammond and Thomas D. Nevitt
Journal of the American Chemical Society 1954 76 (16), 4121-4123
DOI: 10.1021/ja01645a020
OK. When I wrote, above, that the addition of HX to alkenes is not stereoselective, I fibbed. The truth is that it can be stereoselective for anti addition if carried out at low temperatures in non polar solvents such as pentane. The proposed mechanism is not a free carbocation but a termolecular transition state involving two equivalents of H-Br. (Interestingly, though, the reaction of H3O+ with the same compound is not stereoselective).
In the strongly polar solvent acetic acid, the reaction results predominantly (but not exclusively!) through the classic carbocation mechanism. - Hydrochlorination of cyclohexene in acetic acid. Kinetic and product studies
Robert C. Fahey, Michael W. Monahan, and C. Allen McPherson
Journal of the American Chemical Society 1970 92 (9), 2810-2815
DOI: 10.1021/ja00712a034
Detailed kinetic studies of the addition of HCl to cyclohexene in acetic acid, discussing a possible third-order mechanism (rate = k[cyclohexene][HX]2). - SPIROANNELATION OF ENOL SILANES: 2-OXO-5-METHOXYSPlRO[5.4]DECANE
Lee, T. V.; Porter, J. R.
Org. Synth. 1995, 72, 189
DOI: 10.15227/orgsyn.072.0189
The first reaction in the above procedure involves two steps – addition of HBr across the double bond and converting the aldehyde to a dimethyl acetal. - Markovnikov’s Rule
Robert C. Kerber
Journal of Chemical Education 2007 84 (7), 1109
DOI: 10.1021/ed084p1109.1
A 2007 missive urging educators and textbook writers to retire the teaching of Markovnikov’s Rule. - A Case Study in Biomimetic Total Synthesis: Polyolefin Carbocyclizations to Terpenes and Steroids
Ryan A. Yoder and Jeffrey N. Johnston
Chemical Reviews 2005 105 (12), 4730-4756
DOI: 10.1021/cr040623l
Hi, about the addition of HBr to alkenes, without peroxide, does the reaction still follow Markovnikov’s rule?
No. In the presence of peroxides, the reaction is anti-Markovnikov selective. See: https://www.masterorganicchemistry.com/2013/04/12/addition-hbr-alkenes-roor-peroxides-free-radical/
What if you have an alkadiene (non- conjugated), for eg. 3- methyl-1,5-heptadiene with 1 mol of hydrogen halide or water/acid, which one of the double bonds would react?
Protonation will occur so as to give the most stable carbocation intermediate. In that case it’s not very clear which one. But what will happen after protonation is that there will be cyclization to give a new ring, followed by addition of halide ion.
Thank you James. But i have a question. Some sources say that the minor product we considered here does not form. So it does form or not?
A small amount of the “anti-Markovnikov” product will always form. A ballpark estimate is 80 / 20 ratio of markovnikov to anti-markovnikov but it can vary considerably.
You are correct. Fixed, finally!
TYPO: In the diagram in section 2, the red and blue labels are the wrong way round.
Does markovnikov cause steric hindrance?
No, it is irrelevant to steric hindrance.
These notes helps me to clear my doubts and also helps me alot in doing my tutorials
Fantastic explanation
I happened to stubble on this website which she is great amazing.! However the last problem of. CH3
CH3-C-CH=CH2
CH3
hydrochlorination rxn contradicts what you write on the next article linked to this one. At the portion of the alky shift you show the H taken from HCl to follow Markovnikov’s; however, not here.. I may be wrong but this example shown above has caused me great confusion with its contradiction on thethe Article of: Markovnikov’s Rule – Why It Works.