Carboxylic Acid Derivatives
Amide Hydrolysis
Last updated: May 28th, 2026 |
Amide Hydrolysis – Conversion of Amides To Carboxylic Acids
In this post we discuss examples and mechanism of acidic hydrolysis of amides, as well as some examples of amide hydrolysis that are unusually “easy”. We also briefly touch on amide hydrolysis under basic conditions.
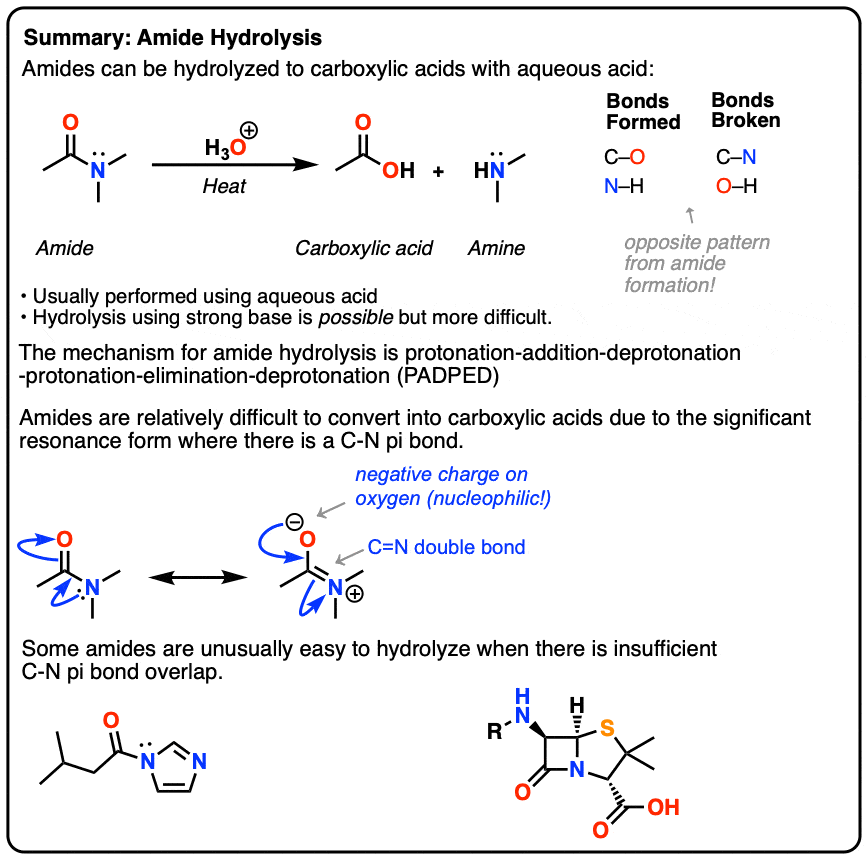
Table of Contents
- Hydrolysis of Amides
- Why Is Hydrolysis of Amides So Difficult Compared To Acid Halides And Esters?
- Amide Hydrolysis Using Aqueous Acid: Mechanism
- What About Basic Hydrolysis of Amides
- Conclusion: Amide Hydrolysis
- Notes
- Supplemental: 3 Amides That Are Unusually Easy To Break
- Quiz Yourself!
- (Advanced) References and Further Reading
1. Hydrolysis of Amides
Amides are carboxylic acid derivatives where the –OH of the carboxylic acid has been replaced by –NH2, –NHR, or –NR2 of an amine. Since the reaction between a carboxylic acid and an amine to give an amide also liberates water, this is an example of a “condensation reaction”. [We discuss the nomenclature and synthesis of amides here].
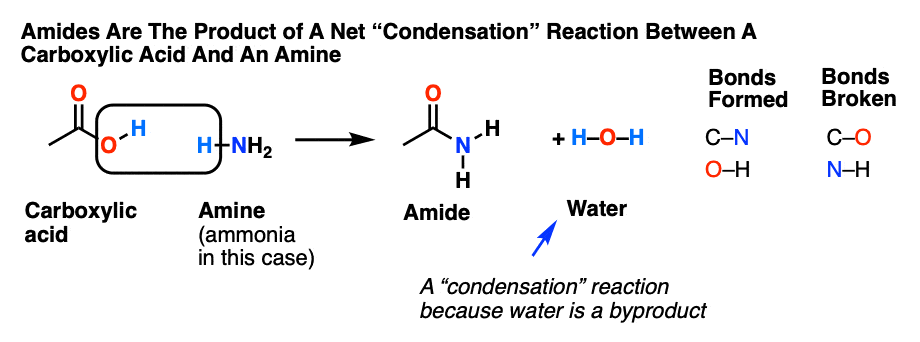
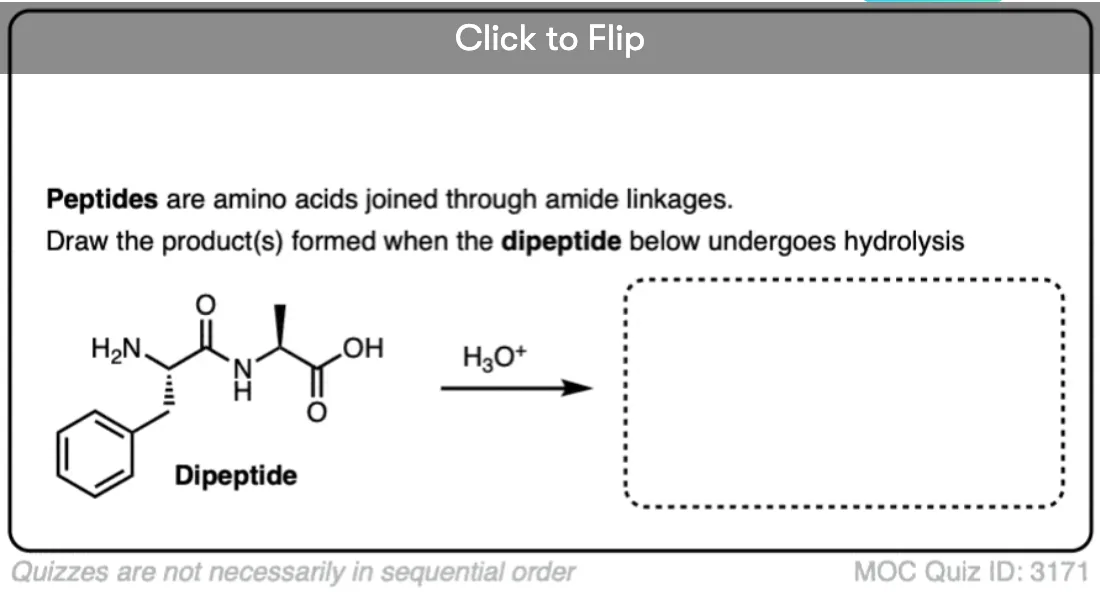
When two amino acids form an amide, we call that species a peptide, and unless you’ve been living under a rock, you’d know that proteins are assembled from amino acids linked by peptide (amide) bonds. Amides are not an easy functional group to break – and a good thing too, since life on Earth is so dependent on them.
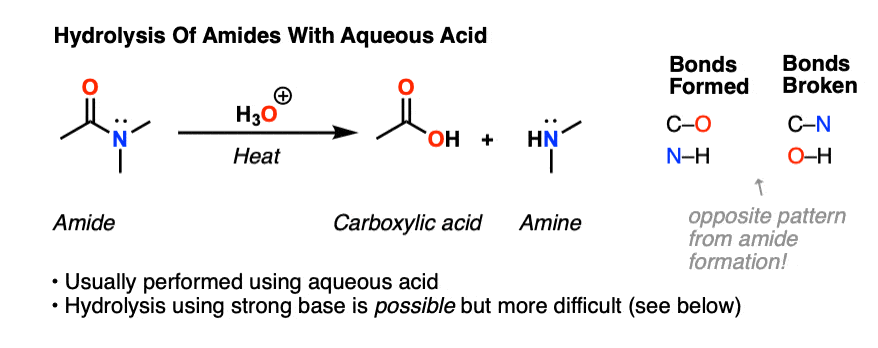
The opposite of a condensation reaction is a hydrolysis reaction. Hydrolysis of amides is typically not an easy thing to do. Typical conditions for hydrolysis of an amide involve heating the amide with aqueous acid for extended periods.
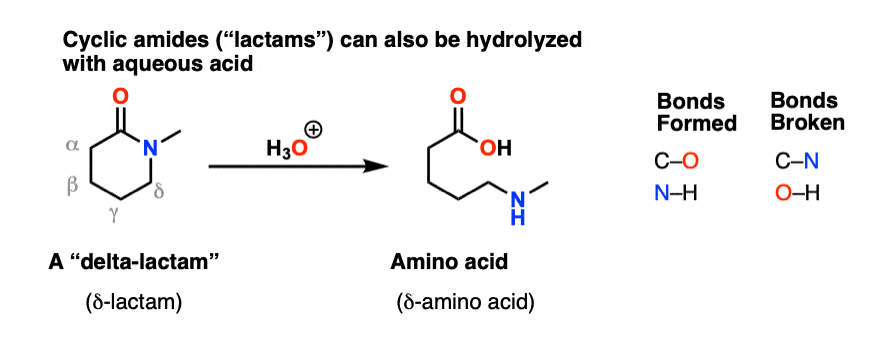
Cyclic amides are called, “lactams”. Just as undoing a belt results in a simple strip of leather, hydrolysis of a cyclic lactam results in a linear amino acid. (The example below is a “delta” amino acid since the amine is a substituent on the fourth carbon down from the carbonyl – not to be confused with the “amino acids” of life, which are “alpha” amino acids).
2. Why Is Hydrolysis of Amides So Difficult Compared To Acid Halides And Esters? Two Reasons
So what makes amides so difficult to break compared to, say, an acid chloride or even an ester.
One key factor is the donating ability of the lone pair on nitrogen. Recall that lone pairs on nitrogen (amines) are less tightly held than lone pairs on (more electronegative) oxygen and thus more available for donation (i.e. more basic). The resonance form with a nitrogen-carbon double bond is thus more significant than the corresponding resonance form for esters. [This theme might be familiar: it’s exactly why –NH2 is a “more activating” substituent than OH in aromatic rings].
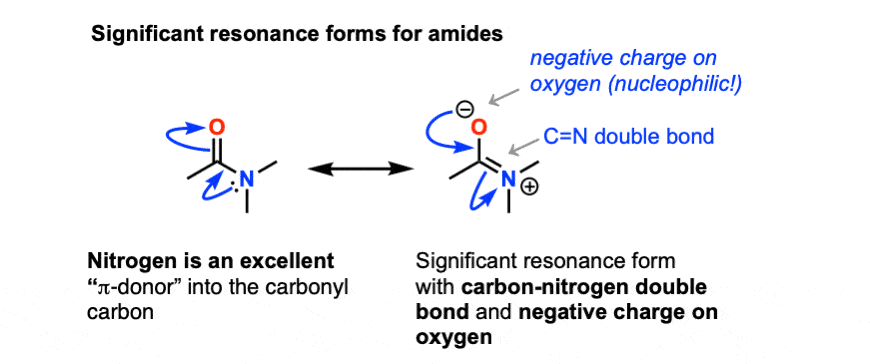
This has several interesting consequences. The first is that the most basic site on an amide is not the lone pair on nitrogen, but instead the oxygen.
Protonation happens on the oxygen first!
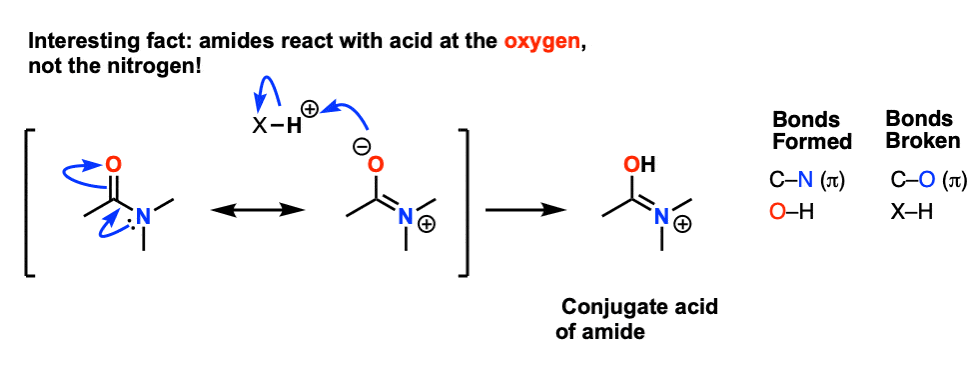
A second interesting consequence is that owing to the importance of that right-hand resonance form, there is significant “double-bond” character in the carbon-nitrogen bond. [In a previous post on Conjugation and Resonance we mentioned that this manifests itself as a barrier to rotation in the C-N bond of about 15-20 kcal/mol ]
3. Hydrolysis of Amides Using Aqueous Acid: Mechanism
All this is to say that performing the hydrolysis of an amide is not nearly so easy as cleaving an acid halide. The mechanism is also not as simple.
So how does the reaction work?
As we noted, the first step is the reversible protonation of the amide on oxygen to give the conjugate acid.
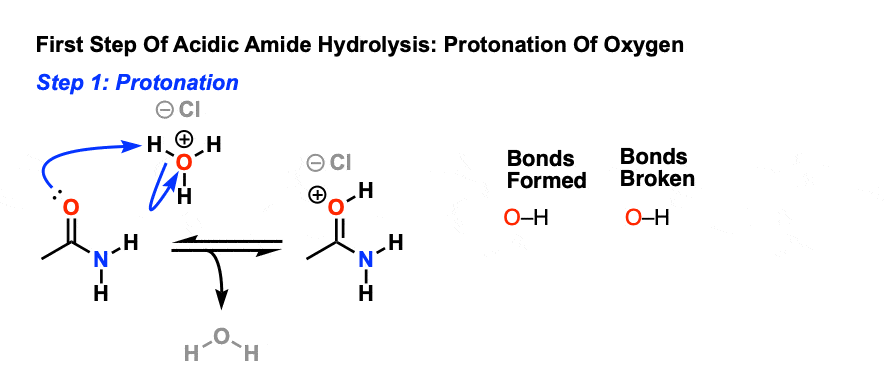
Protonation of the carbonyl oxygen makes the carbonyl carbon a better electrophile, since the C-O pi bond is weakened and the resonance form with a carbocation on carbon becomes more significant.
The next step is then addition of a nucleophile (water, which is either solvent or co-solvent) forming a new C-O bond and breaking the C-O pi bond.
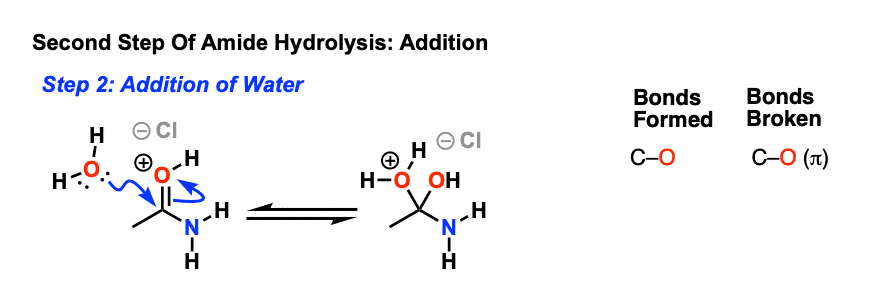
This results in a new species with a positive charge on oxygen. In the next step, a proton is shuttled over to the nitrogen atom through deprotonation of oxygen and protonation of nitrogen. [ deprotonation – protonation, or just “proton transfer”]
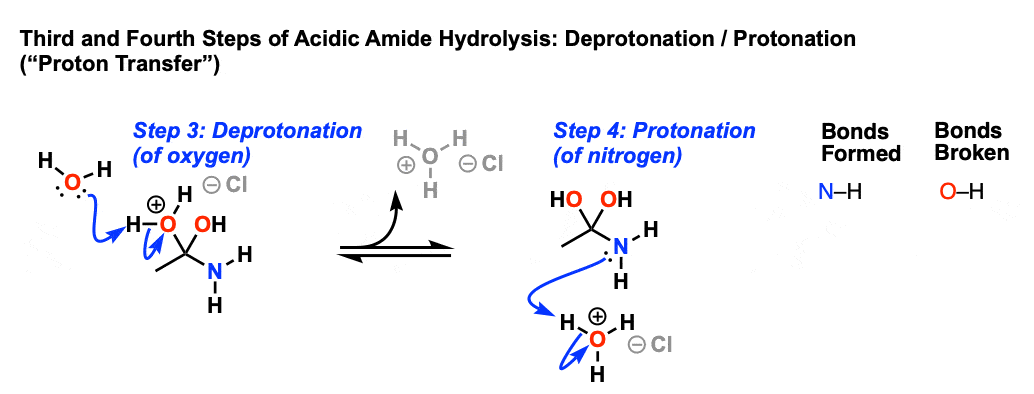
The resulting positively charged nitrogen species is now a significantly better leaving group, since the leaving group will be HNR2 (a weak base) instead of (–)NR2 ( a very strong base). Hence in the next step elimination occurs, forming a new c-o pi bond and breaking C-N.
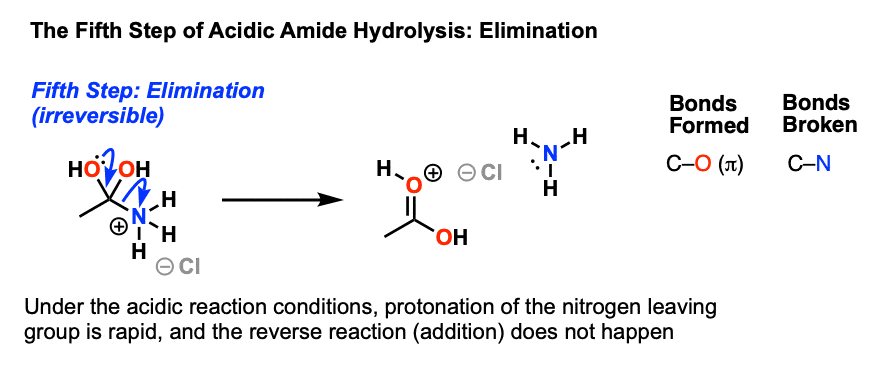
This results in a positively charged carboxylic acid derivative, which is then deprotonated to give the neutral carboxylic acid, completing the hydrolysis of the amide.
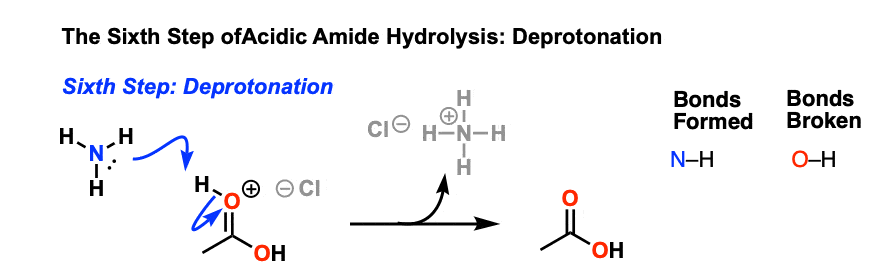
This six-step mechanism (protonation, addition, deprotonation, protonation, elimination, deprotonation) might seem vaguely familiar. It is the exact sequence of steps in Fischer esterification and various other mechanisms, going by the acronym PADPED. [See Making Music With Mechanisms]
All the steps of the process are in equilibrium until the elimination reaction occurs. Once carbon-nitrogen bond has been broken, addition is extremely unlikely since the amine is present as its conjugate acid and can’t act as a nucleophile.
For your average amide, that’s essentially all there is to amide hydrolysis. Obviously for a primary amide, the leaving group will be NH3, and for a secondary amide it will be RNH2.
4. What About Basic Hydrolysis of Amides?
So that’s acidic hydrolysis. What about basic hydrolysis?
It can be done, but it’s typically not easy. If brute force is insisted upon, it’s possible. Hydrolysis of amides with base requires prolonged heating.
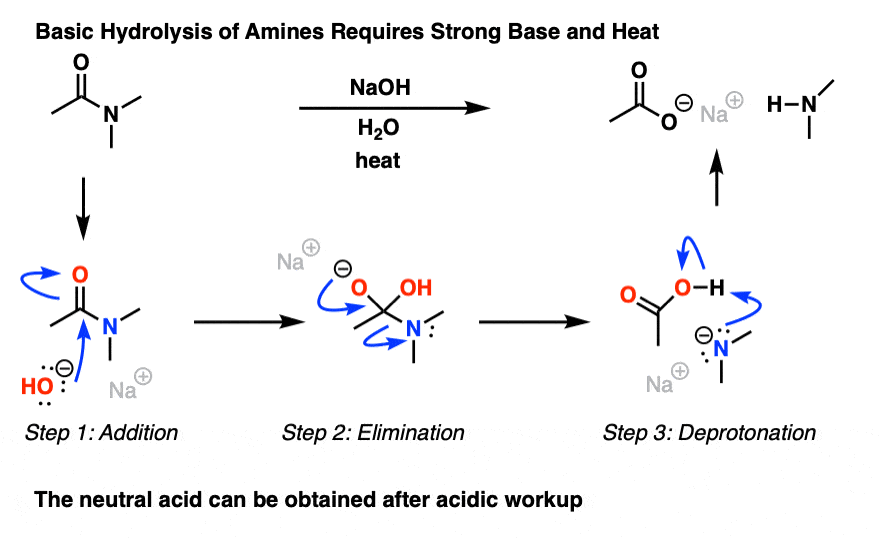
The whole problem is that in order for a substitution reaction to occur (whether it be SN2 or acyl substitution) you need a decent leaving group. Since it is such a strong base, a deprotonated amine (confusingly also called an “amide”, or sometimes “metal amide base”) is pretty much the opposite of a decent leaving group. So even with a strong base like potassium hydroxide and lots of heat, cleaving an amide can be difficult. [Note 1]
5. Summary: Hydrolyzing Amides To Carboxylic Acids With Acid Or Base
Acidic hydrolysis of amides is one of those “meat and potatoes” reactions of chemistry that are essential to know and understand. One key to thoroughly understanding the mechanism is to break the reaction down to its six steps (PADPED) and compare it to reactions that share this core mechanistic pathway (e.g. the Fischer Esterification, hydrolysis of esters, and more).
Make sure you can draw the product for the hydrolysis of a cyclic amide (lactam) since reactions of cyclic molecules (and their reverse, intramolecular reactions) are common fodder for exams.
This reaction comes up later in the Strecker synthesis of amino acids, which begins with the addition of cyanide ion to an imine, followed by hydrolysis of the nitrile to give the carboxylic acid.
Thanks to KG for assistance with the figures in this post!
Notes
Note 1. Some studies suggest that breakage of the C-N bond does not occur until the second OH group is deprotonated.
Supplemental: 3 Amides That Are Unusually Easy To Break
Amides That Are Unusually Easy To Break (1) – Acylimidazole
As we said, amides tend to be difficult to cleave. However it’s worth looking at some exceptions that help to illustrate the key points here.
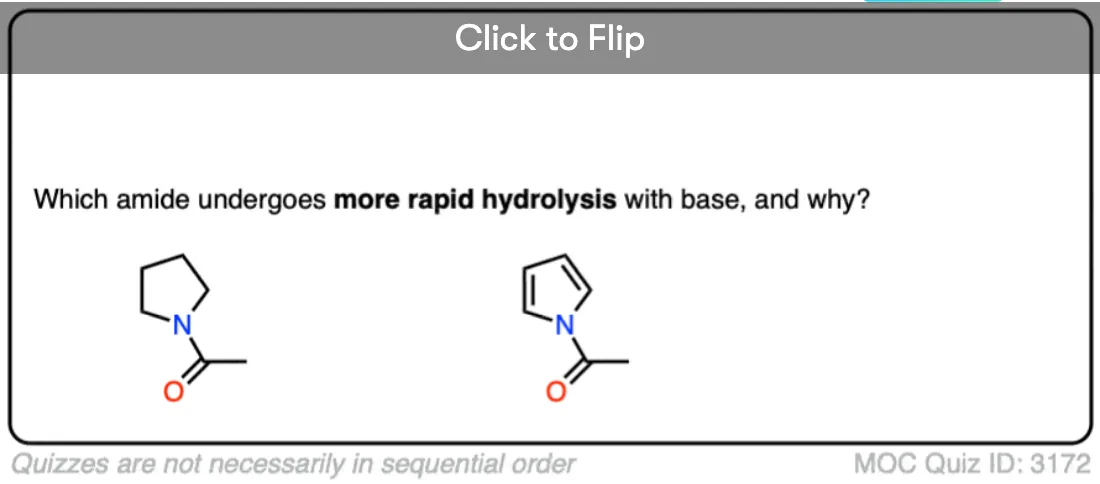
One particularly easy amide to break is acyl imidazole. There’s still a C-N bond, and there’s still a lone pair on nitrogen.
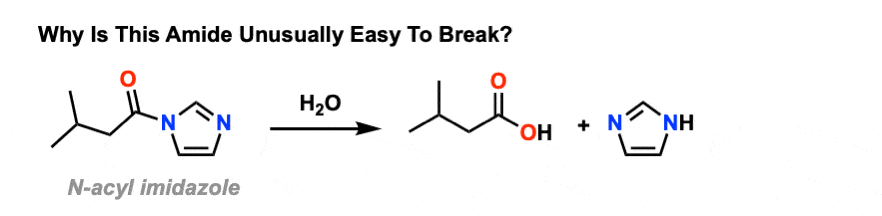
So why is it so easy to break?
Think about the resonance forms. What do you notice about imidazole in the resonance form on the left versus the resonance form on the right?
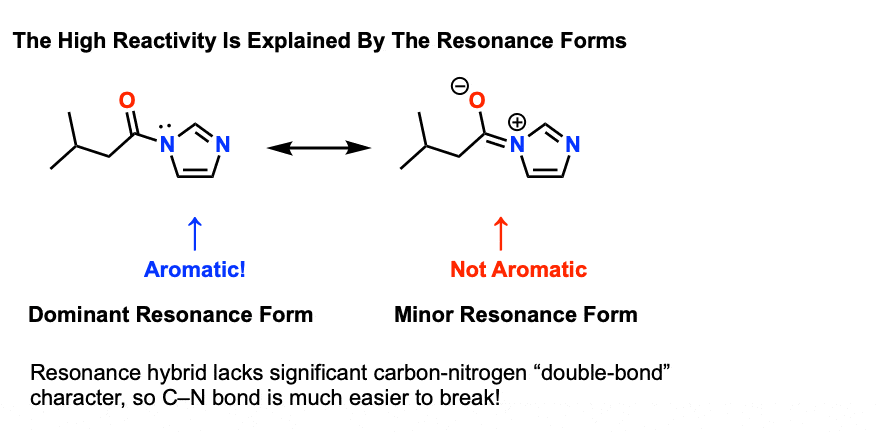
In the resonance form on the [left], the imidazole is aromatic. In the resonance form on the right, the one with partial C-N double bond character, that aromaticity is lost.
This is not unique to N-acylimidazole. It’s also true for N-acylpyrrole, N-acylindole, and other species where the lone pair is “tied up” in an aromatic ring.
Amides That Are Unusually Easy To Break (2) – Beta-Lactams
As described in The Enchanted Ring, MIT chemist John Sheehan and his research group had a hell of a time synthesizing penicillin. The trouble was as soon as they would form the 4-membered amide ring (a “beta lactam”) using conventional conditions the damn thing would fall apart. Eventually Sheehan’s group developed DCC (and later EDC) as a very mild method for forming amide linkages and the problem was solved.
The beta-lactam is unusually easy to break for two reasons. First, and most obvious, is the fact that the resonance form where there is a carbon-nitrogen double bond is in a 4-membered ring, and a four membered ring with a double bond (e.g. cyclobutene) adds even more strain to the system. This minimizes the importance of the resonance contributor with the C=N double bond. A second, more subtle reason is that the sp3 hybridized carbon on the ring junction (adjacent to the nitrogen) imparts a slight pucker to the nitrogen, so that orbital overlap is even more difficult than in a linear amide.
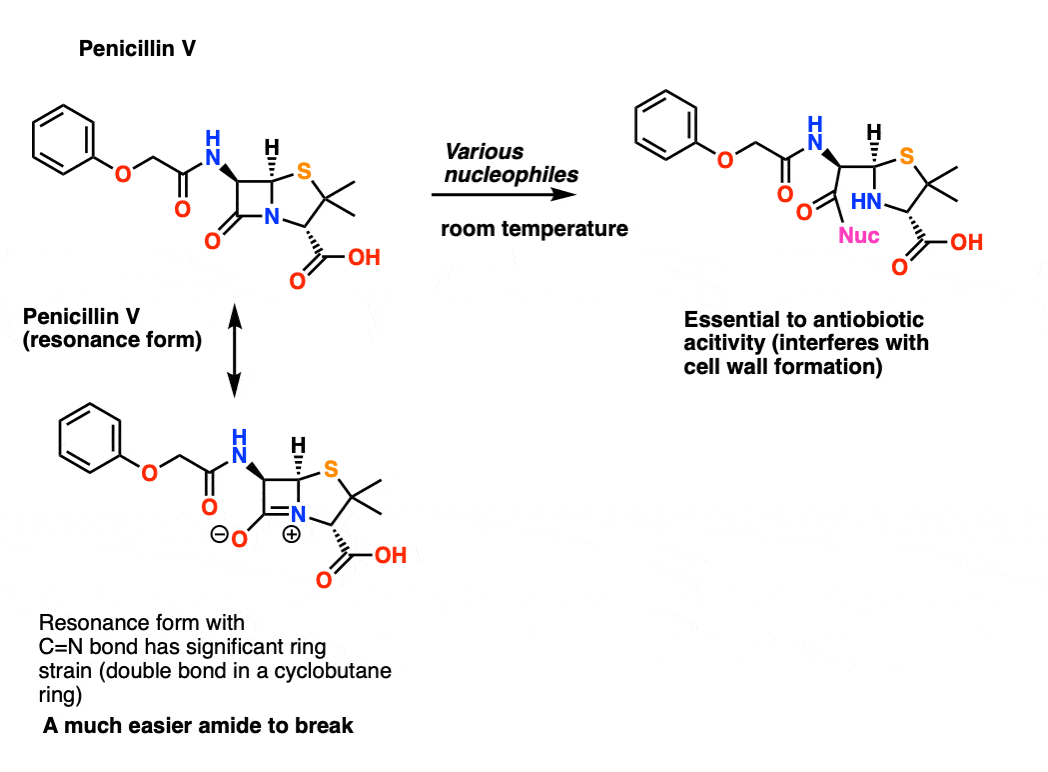
Since orbital overlap is bad, the carbon-nitrogen bond lacks partial double-bond character, and it’s easy to break.
Amides That Are Unusually Easy To Break (3) – Quinuclidine Amide
A related example is this “bridgehead” amide quinuclidinone. While it might not look so strange at first glance, when you build a model you see that the nitrogen lone pair is pointing out at a weird angle that prevents overlap with the adjacent carbonyl. The crystal structure on the right (from this study by Prof. Brian Stoltz at Caltech) makes the lack of orbital overlap even more obvious.
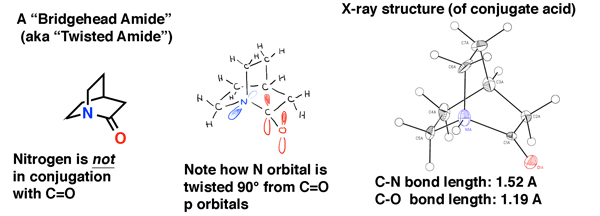
Remember Bredt’s rule about how bridgehead alkenes are unstable? The same is true of amide nitrogens at a bridgehead. In order for “partial double bond character” to be present in that resonance form, there has to be orbital overlap, and as Bredt’s work showed, for reasonably small ring sizes that orbital overlap is extremely weak.
Quiz Yourself!
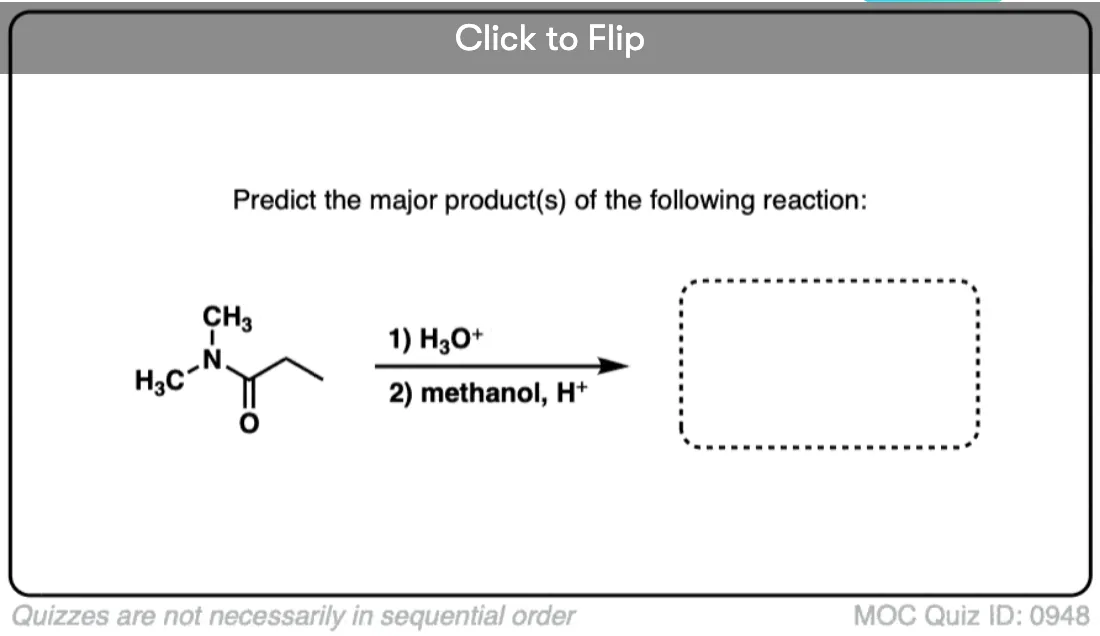
Become a MOC member to see the clickable quiz with answers on the back.
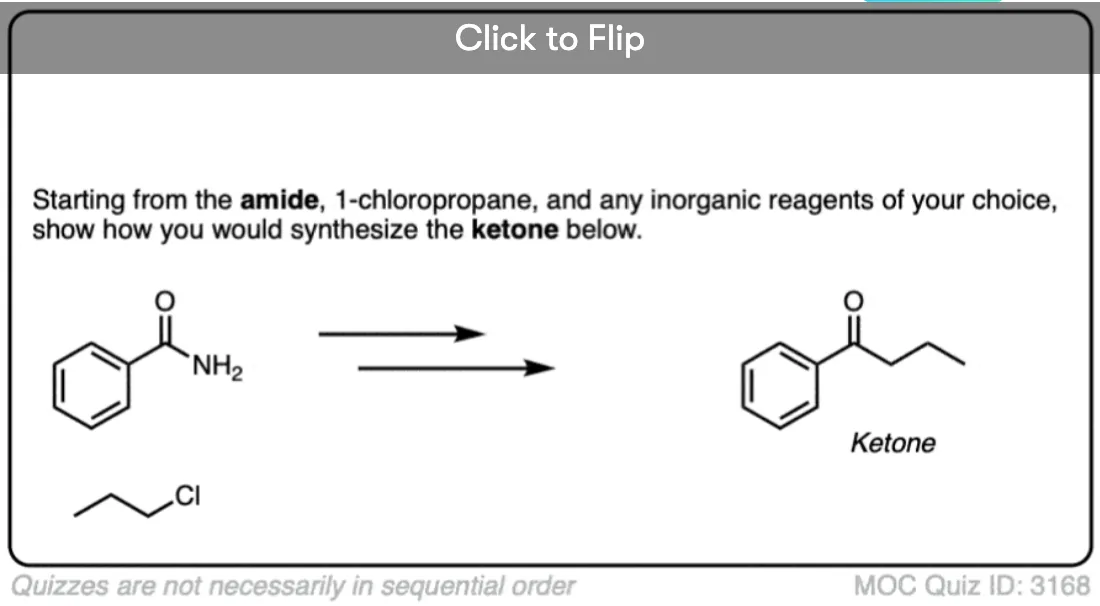
Become a MOC member to see the clickable quiz with answers on the back.
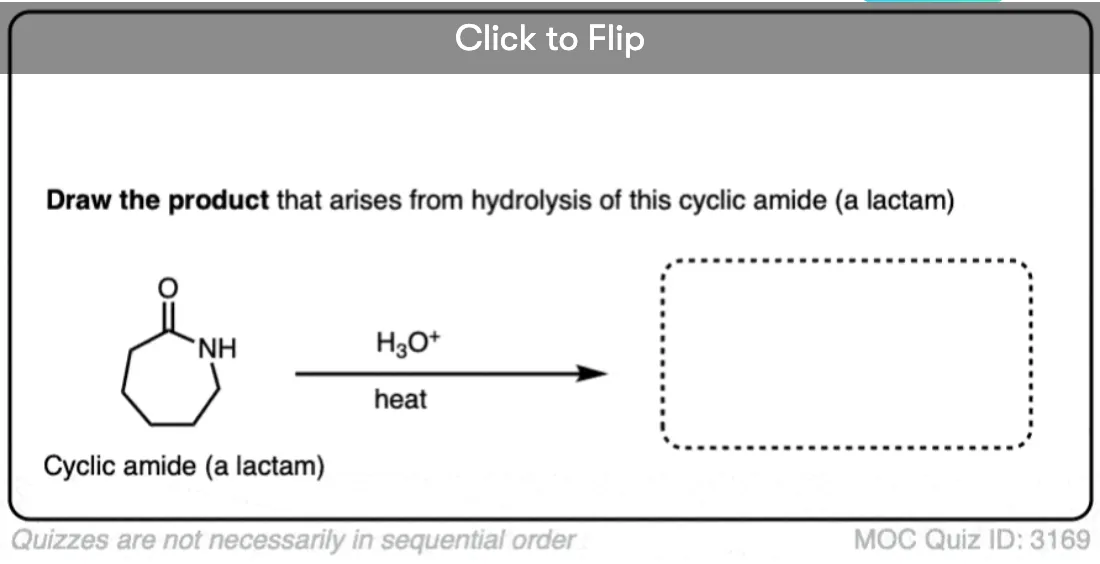
Become a MOC member to see the clickable quiz with answers on the back.
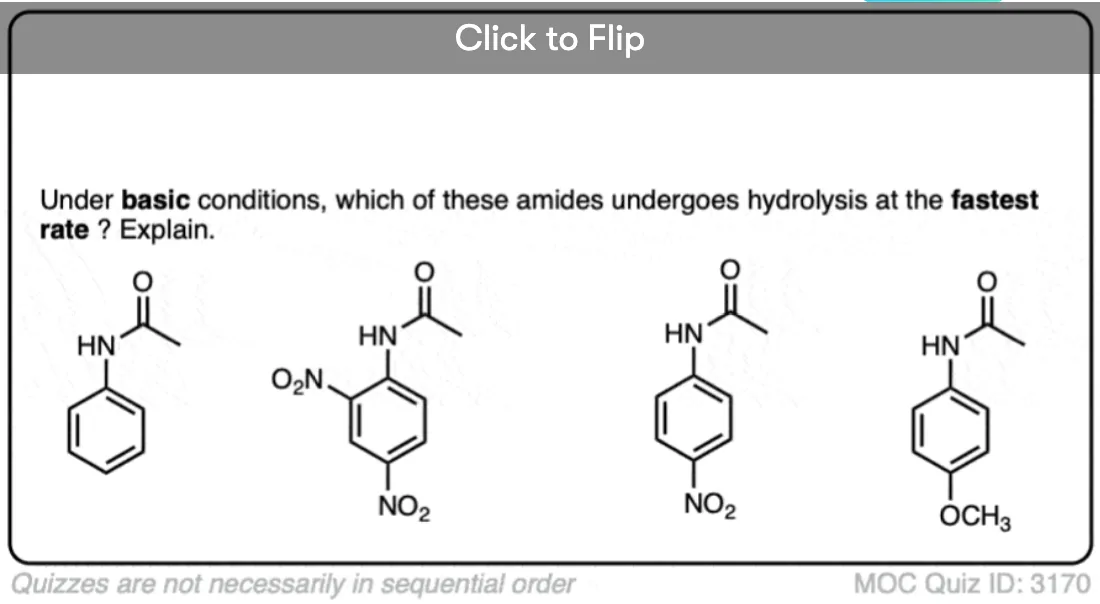
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading
- α-AMINODIETHYLACETIC ACID
Steiger, Robert E.
Org. Synth. 1942, 22, 13
DOI: 10.15227/orgsyn.022.0013
For an example of acidic amide hydrolysis, this procedure in Organic Syntheses is fairly typical. A primary amide is refluxed in concentrated HCl for 2.5 hours to obtain the carboxylic acid. - Catalytic Efficiencies in Amide Hydrolysis. The Two-Step Mechanism
Richard L. Schowen, H. Jayaraman, and Larry Kershner
Journal of the American Chemical Society 1966 88 (14), 3373-3375
DOI: 10.1021/ja00966a034
Mechanistic study of basic hydrolysis of amides supports a two step addition-elimination mechanism. - Synthesis and structural analysis of 2-quinuclidonium tetrafluoroborate
Kousuke Tani & Brian M. Stoltz
Nature 441, pages 731–734 (2006)
DOI: 10.1038/nature04842
Synthesis and structure of an extremely unstable “twisted” amide by Stoltz and Tani, complete with crystal structure.
Thank you James sir! Very helpful!
Hi James,
I do not understand what that last note means. Would you explain that please