Dienes and MO Theory
Reactions of Dienes: 1,2 and 1,4 Addition
Last updated: May 28th, 2026 |
Kinetic Versus Thermodynamic Control In Addition of HBr to Dienes: 1,2- and 1,4- Addition
In today’s post we’ll discuss 1,2- and 1,4- addition to dienes – specifically, the addition of strong acid such as HBr.
- When a diene undergoes reaction with a strong acid like HBr, protonation results in a resonance-stabilized carbocation
- The resonance-stabilized carbocation can undergo attack at two possible positions.
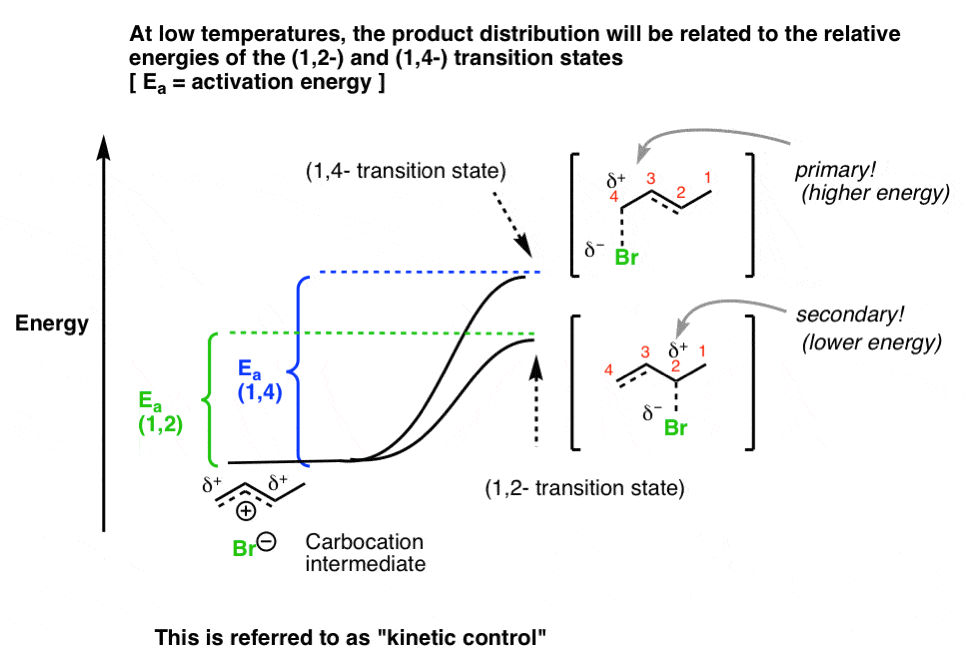
- When the reaction is conducted at low temperatures, the reaction is irreversible and the major product will be the one with the lowest-energy transition state, which is the carbon best able to stabilize positive charge.
- This is referred to as running the reaction under kinetic control.
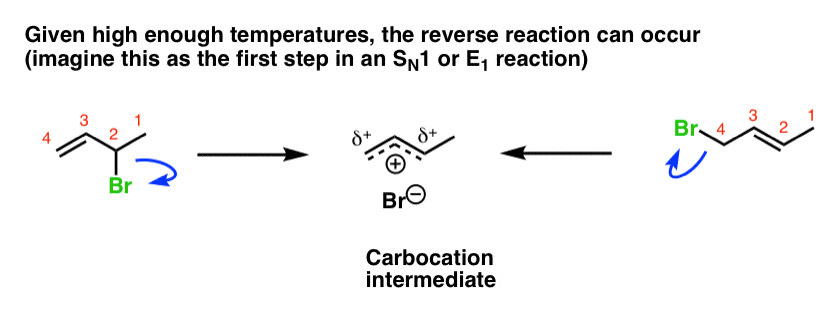
- When the reaction is conducted at higher temperatures, the reaction is reversible and the major product will be the one which is most thermodynamically stable, which is generally the most-substituted alkene.
- This is referred to as running the reaction under thermodynamic control.
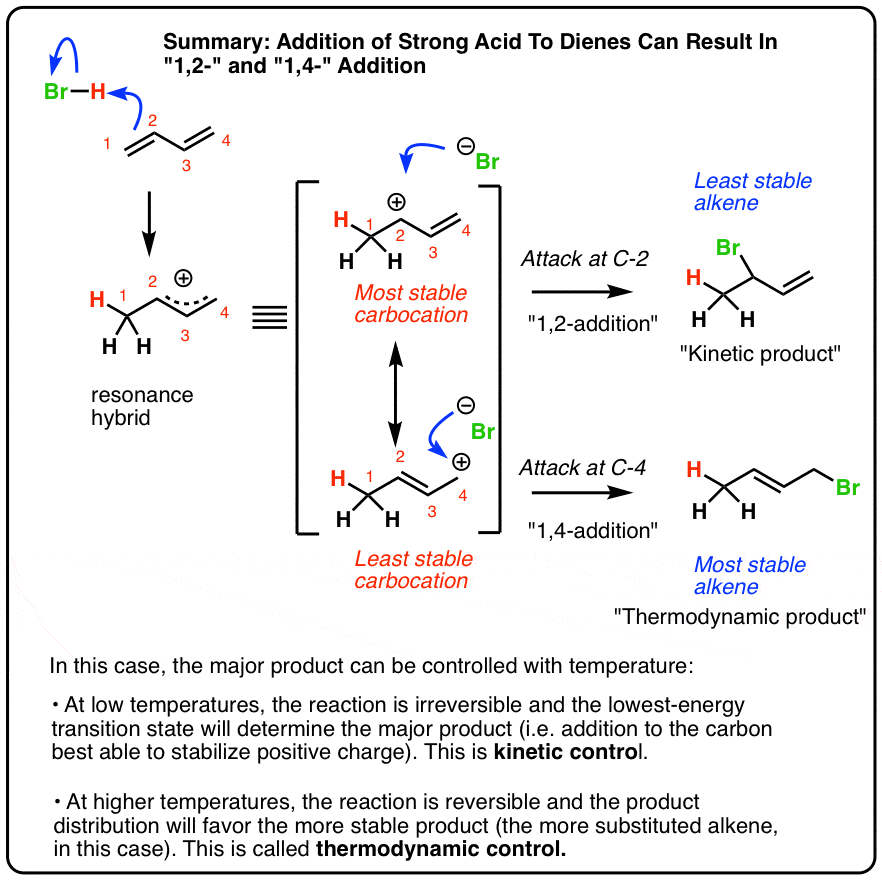
Table of Contents
- Addition To Alkenes, Revisited
- Reaction Of Butadiene With Acid Gives “1,2-Addition” And “1,4-Addition” Products
- The “1,4 Addition” Product Of Acids Adding To Butadiene
- Protonation Of Butadiene Gives A Resonance-Stabilized Carbocation
- The Effect Of Temperature: Low Temperatures Give “1,2” Addition To Butadiene
- Higher Temperatures Give More Of The “1,4” Product
- With Butadiene, The “1,4” Product Is More Stable Because It Has A More Substituted Double Bond
- “Kinetic Control” vs “Thermodynamic Control”: At Low Temperatures The Reaction Is Irreversible And Products Are Determined By Relative Rates
- Thermodynamic Control: At Higher Temperatures The Reaction Is Reversible And Product Distribution Is Determined By Stability
- Using An Energy Diagram To Understand Thermodynamic Versus Kinetic Control
- A Parting Word Of Warning: The “1,4-Product” Is Not Always The “Thermodynamic” Product
- Other Reactions Can Give 1,2- and 1,4- Additions As Well
- Notes
- Quiz Yourself!
- (Advanced) References and Further Reading
1. Addition To Alkenes, Revisited
Waaay back in the day we talked about addition reactions of alkenes. Remember Markovnikov’s rule in the addition of electrophiles to alkenes?
For example, take an alkene like 1-butene, and add HBr. What happens?
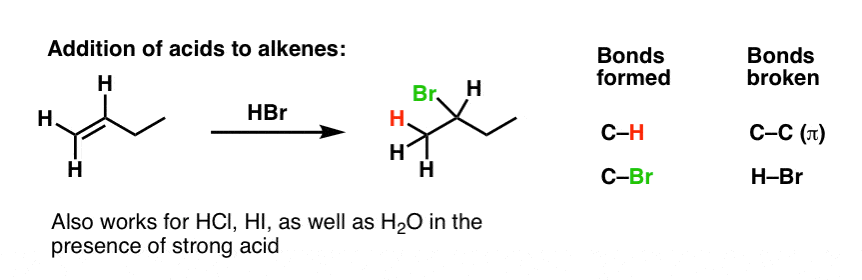
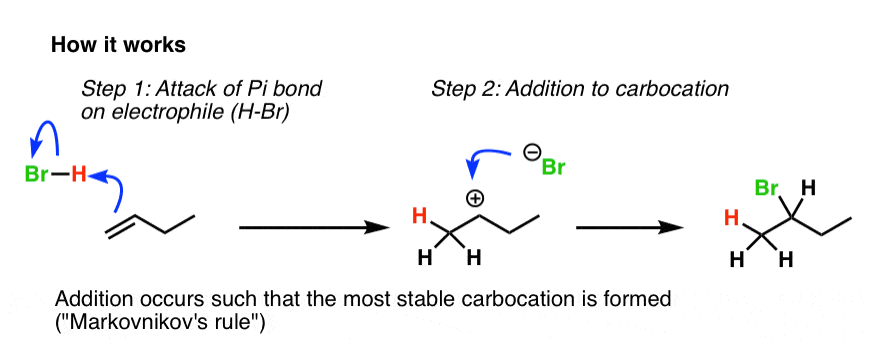
An addition reaction occurs! (Break C-C pi, and form adjacent C-H and C-Br bonds).
Bromine adds to the most substituted carbon of the alkene, and hydrogen adds to the least substituted carbon. That’s due to carbocation stability: the reaction proceeds through the most stable carbocation, which happens to be the most substituted.
2. Reaction Of Butadiene With Acid Gives “1,2-Addition” And “1,4-Addition” Products
All well and good. But since we’ve been discussing conjugation and resonance, let’s throw in an additional wrinkle.
What happens when we try the same reaction on a diene? Butadiene, for example.
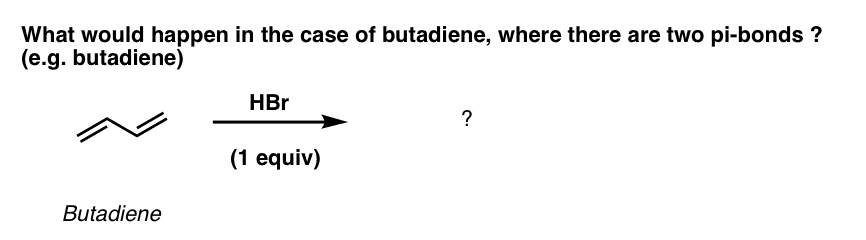
I’ll tell you. You get two products! (But the second one might not be what you think it is).
Here’s what they look like:
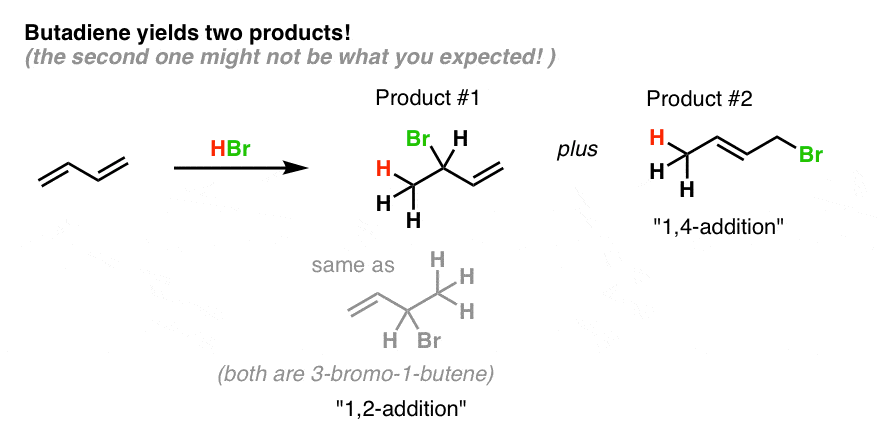
Product #1 is essentially the same product that we saw in the 1-butene example: H and Br are added across two consecutive carbons of a double bond. Note that since butadiene is symmetric, the same product is formed regardless of which double bond participates! (we get 3-bromo-1-butene either way).
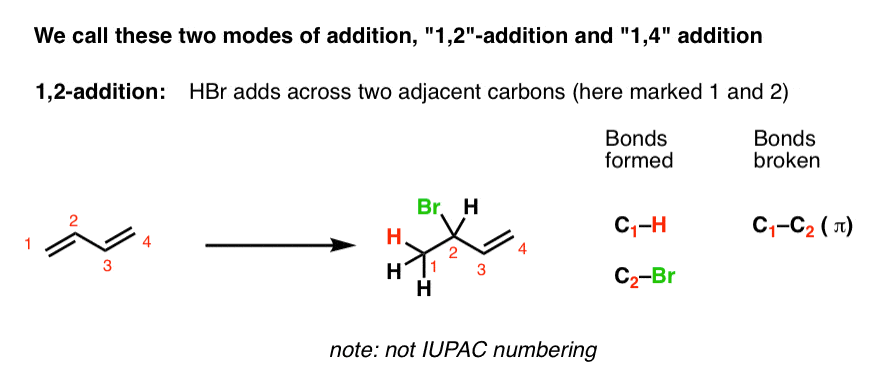
Since addition occurs across two consecutive carbons, we often call this “1,2 addition”.
3. The “1,4 Addition” Product Of Acids Adding To Butadiene
In contrast, Product #2 shows the result of adding H and Br across four conjugated carbons. All four carbons participate in the reaction. A new C-H single bond has formed on one end of the diene (C-1), and C-Br formed on the other end (C-4). Note that the C1-C2 and C3-C4 pi bonds are broken, and we’ve formed a new pi bond between C2 and C3.
We call this “1,4 addition”.
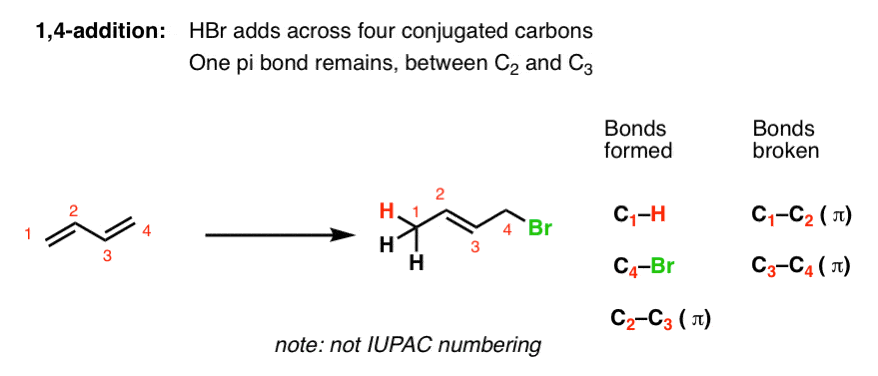
[Note: to simplify the discussion here, I’m choosing to ignore double bond isomers (i.e. E and Z) in this analysis. In the lab, the 1,4- example above will exist as a mixture of (mostly) E product with a small amount of the Z. ]
So why does this “1,4 addition” happen in the first place? What’s different about butadiene, as opposed to 1-butene where only “1,2-addition” was possible?
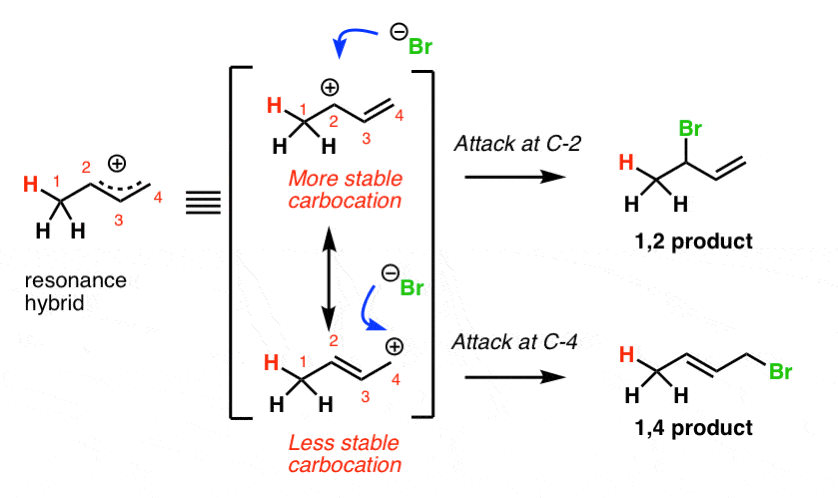
4. Protonation Of Butadiene Gives A Resonance-Stabilized Carbocation
The first thing to notice is that the initial protonation of butadiene gives a resonance-stabilized carbocation. See how protonation of C-1 gives a carbocation that has two important resonance forms?
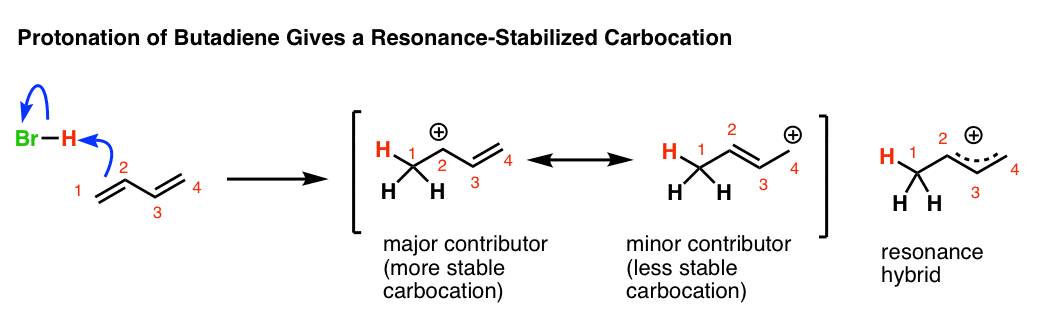
In the case of butadiene,
- the major contributor to the resonance hybrid will be the resonance form where the carbocation is on the more substituted carbocation (C2)
- the minor contributor will be the resonance form where the carbocation is on the less substituted carbocation (C4).
Attack of the nucleophile (Br–) at the C2 position of the hybrid will lead to the 1,2-product.
Attack of Br– at the C4 position of the resonance hybrid will lead to the 1,4-product.
{kind=link}
[Note: never forget that resonance forms are not in equilibrium with each other. It’s understandable to casually say that “Br(–) will attack the top resonance form with the more stable carbocation” so long as you remember that the true structure of the molecule is a hybrid of the two resonance forms.] In the footnote, I’m including perhaps a more correct way to show formation of the bottom resonance form.
5. The Effect Of Temperature: Low Temperatures Give “1,2” Addition To Butadiene
Here’s the big question: which one of the two products will be major? The 1,2-addition product or the 1,4-addition product?
We’re so used to seeing “Markovnikov addition” in alkenes (where addition occurs to the most stable carbocation) that it seems intuitive that the 1,2-addition product would be dominant.
And indeed, at low temperature, 1,2 addition to butadiene is favoured. Here’s a literature example.
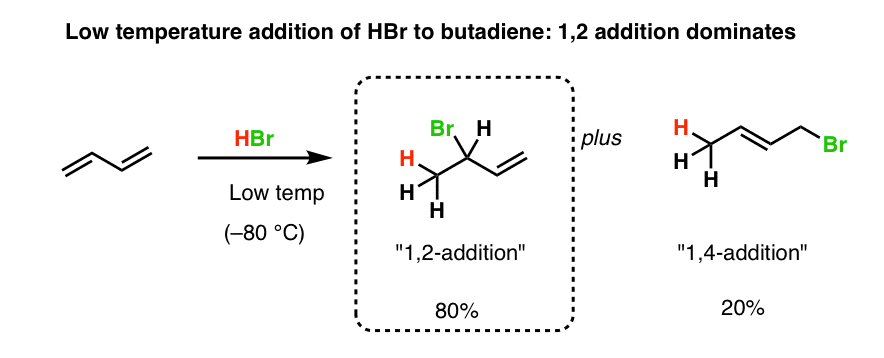
6. Higher Temperatures Give More Of The “1,4” Product
Interestingly, however, as the temperature is increased, the amount of 1,4 product increases.
At room temperature, the ratio of 1,2- and 1,4- addition is 45:55 .
At 40 degrees Celsius the 1,4- product is dominant (about 80%).
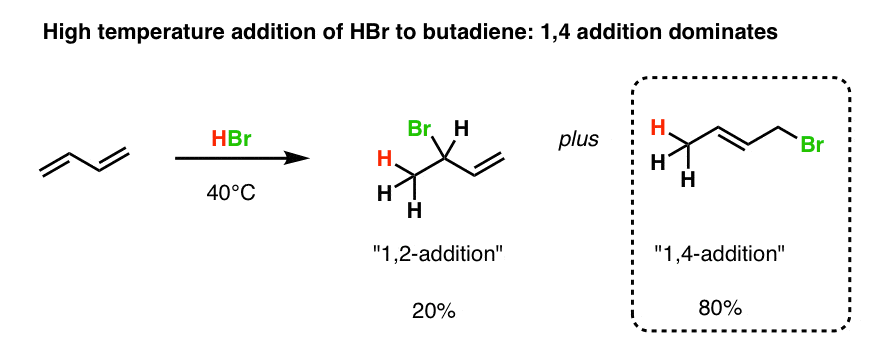
7. With Butadiene, The “1,4” Product Is More Stable Because It Has A More Substituted Double Bond
What’s going on here? What structural features are present that could possibly make formation of the 1,4 product more favourable than the 1,2 product, even though it goes through a “less stable” carbocation?
The answer lies in the substitution pattern of the double bond.
Remember Zaitsev’s rule? Same deal. The 1,4 product is more stable because it is a more substituted double bond.
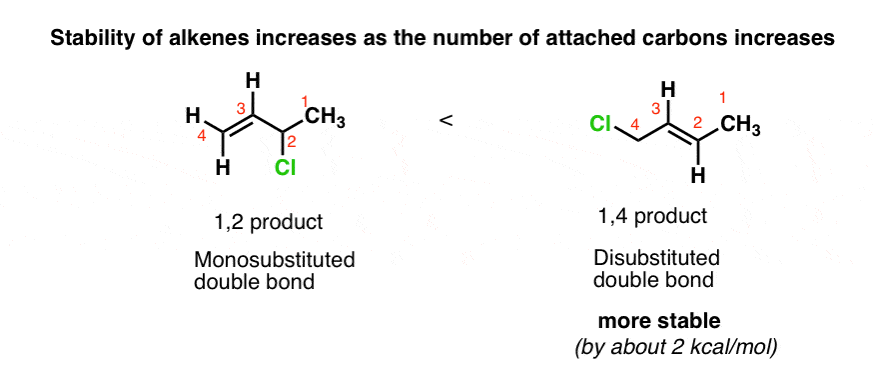
The 1,4 product has a di-substituted double bond, whereas the 1,2-product has a mono-substituted double bond. Generally speaking, double bond stability increases as the number of carbons directly attached to the double bond is increased. (See article: Alkene Stability)
So why would 1,4 be more favoured under conditions of higher temperature, and the 1,2 be favoured under conditions of lower temperature?
8. “Kinetic Control” vs “Thermodynamic Control”: At Low Temperatures The Reaction Is Irreversible And Products Are Determined By Relative Rates
At low temperatures, the differentiating factor is the relative energies of the transition states leading to the products. The 1,2 product has a lower-energy transition state, owing to the fact that charge is more stable on the more substituted carbon. The difference between the energies of these transition states will determine the product ratio.
A quick analogy. Imagine you’re hungry, and you only have $5 in your pocket. In a choice between McDonalds and Applebee’s, the only accessible option is McDonalds – even if, for some reason, you like Applebee’s a lot more.
Let’s sketch this out. The carbocation intermediate can pass through the two different transition states that lead to the 1,2- and 1,4- products, respectively. If we choose a temperature low enough, then the product distribution will reflect the difference in energy between the two activation energies Ea (1,2) and Ea (1,4). So long as the reaction is not reversible, the product with the lower energy transition state will dominate. This is called “kinetic control”.
9. Thermodynamic Control: At Higher Temperatures The Reaction Is Reversible And Product Distribution Is Determined By Stability
At higher temperatures, the reaction has the potential to be reversible. [Note 2] In this case, this means is that both the 1,2- and 1,4- products can ionize (think of the first step in the SN1 reaction), reforming the carbocation intermediate.
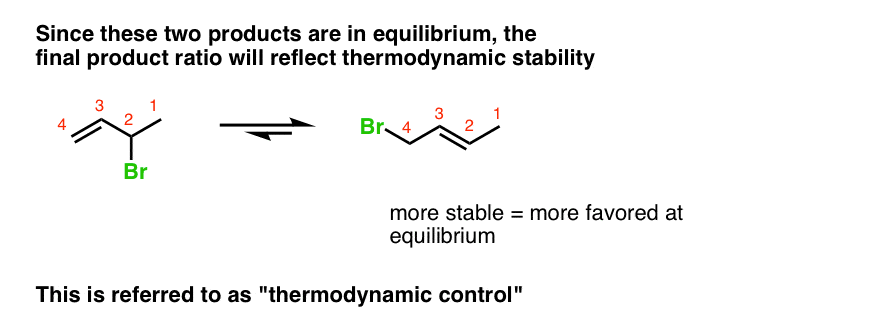
This sets up an equilibrium. The product ratio will now reflect the relative stabilities of the 1,2- and 1,4- products, not the transition states leading to their formation. In the case of butadiene, since the 1,4- product is more stable (it has a disubstituted double bond) it will be the dominant product at higher temperatures.
This is referred to as “thermodynamic control“.
Math interlude: recall that ΔG = –RT lnK
A difference of 1 kcal/mol in stability doesn’t sound like much, but it translates into a 83:17 ratio of products at equilibrium at room temperature.
Continuing our analogy: with enough money in your pocket, the decision where to eat lunch is more a function of how much you like the overall McDonalds vs. Applebee’s experiences, not how much they cost.
10. Using An Energy Diagram To Understand Thermodynamic Versus Kinetic Control
Drawing up the reaction energy diagram can be helpful to understand kinetic and thermodynamic control. This post here goes into more detail, but we’ll repeat the basics here.
The reaction energy diagram for the addition of HBr to butadiene looks like this:
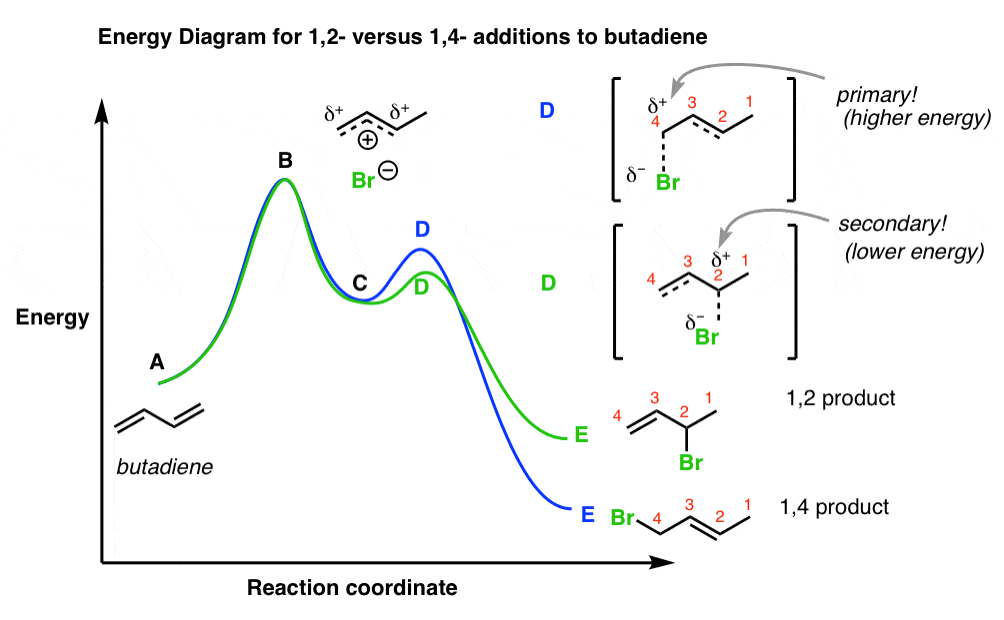
Point A is the starting butadiene, and point B is the transition state for addition of H to butadiene.
The important part to pay attention to is the “local minimum” C, the resonance-stabilized carbocation.
As we mentioned previously, going from intermediate C to transition states D and D represent the energy pathways for 1,2- and 1,4- addition, respectively.
Hence the activation energies for the forward reactions is equal to the difference in energy between D and C (for 1,2-addition) and D and C (for 1,4-addition). We saw that 1,2-addition has a lower activation energy.
Now let’s look at the reverse reaction.
Points E and E represent the energies of the 1,2- and 1,4- products.
The activation energy for the reverse reaction is the difference in energy between E and transition state D, and E and transition state D, respectively.
It should be clear from this diagram that the activation energies for the forward reaction (going from C through transition states D and D give E and E, respectively) are much lower than the activation energies going in the reverse reaction (i.e. from E and E through transition states D and D back to carbocation C ).
- So if we keep the temperature low, we favor the forward reaction and hinder the reverse, and obtain the kinetic product.
- If the temperature is raised, the reverse reaction becomes energetically accessible, and equilibrium is established. We will then obtain the thermodynamic product.
11.A Parting Word Of Warning: The “1,4-Product” Is Not Always The “Thermodynamic” Product
Let’s summarize. Understanding the following two factors is key to correctly answering exam problems.
The relative importance of the carbocation resonance forms- The relative stabilities of the double bond products.
In the case of butadiene, it is true that the 1,2 product was formed through a more stable carbocation (kinetic product), and the 1,4 product had a more stable double bond (thermodynamic product).
But it will not always be true for all dienes!!
Here are three examples that will help you think through the main issues (answers in the next post).
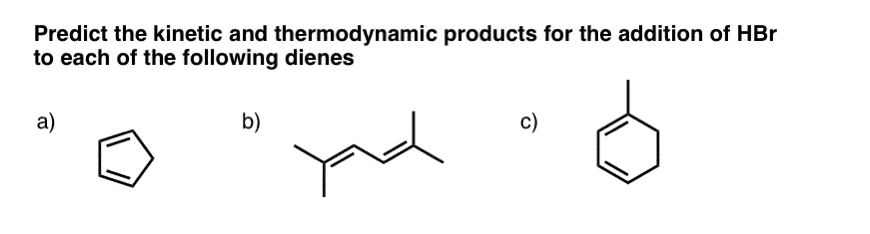
12. Other Reactions Can Give 1,2- and 1,4- Additions As Well
This post is long enough, but I would be remiss if I failed to note that 1,2 and 1,4 additions to dienes are also possible for a few other classes of reaction.
- Addition of HBr to dienes under free-radical conditions (e.g. HBr + peroxides)
- Addition of Br2 (and Cl2) to dienes.
Note that similar issues will arise (i.e. stability of a reactive intermediate versus stability of the final product). We’ll go into more detail when the time comes.
Many thanks to Tom Struble for help with preparing this post.
Notes
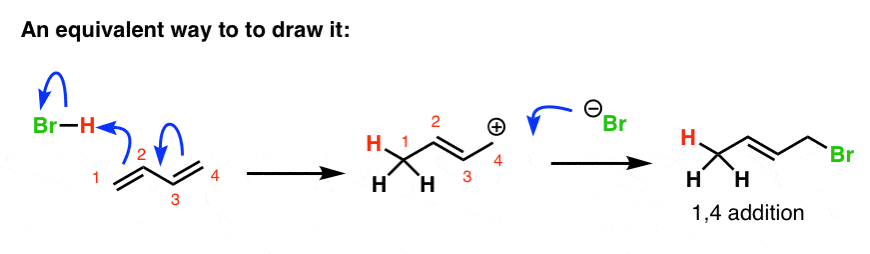
Note 1. All the textbooks I consulted showed diagrams similar to what I drew above, with Br(-) attacking different resonance forms. Perhaps a more correct way to draw the formation of the carbocation at the 4-position is to show it like this.
Quiz Yourself!
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
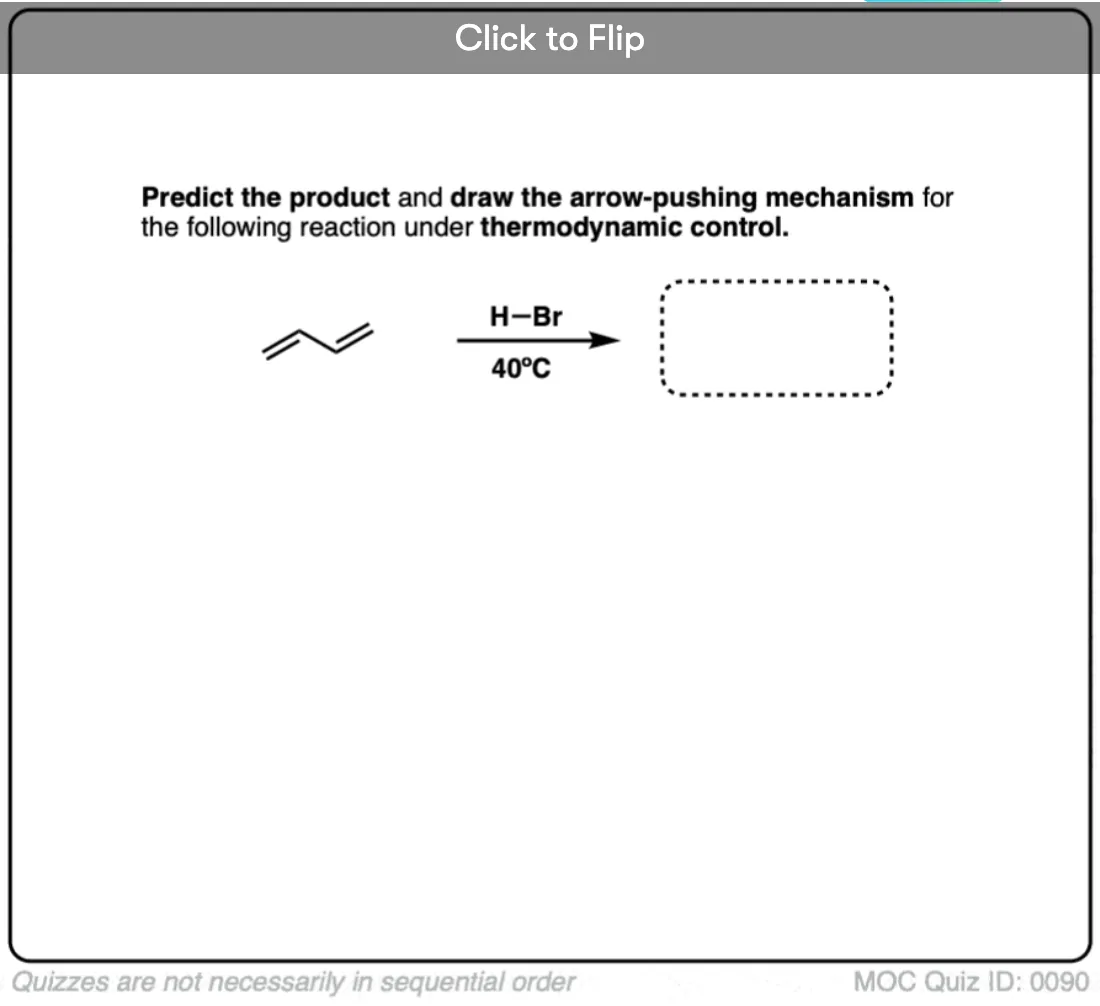
Become a MOC member to see the clickable quiz with answers on the back.
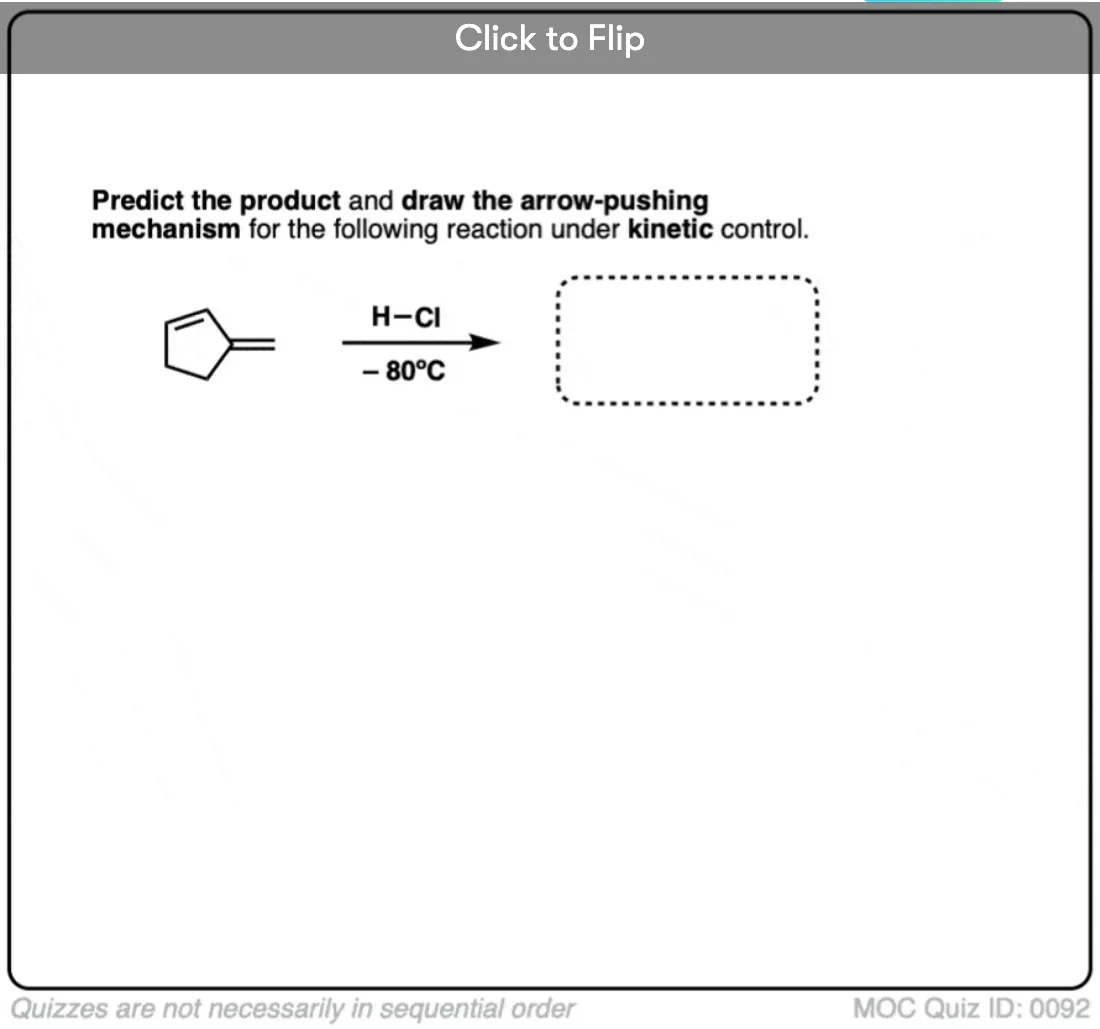
Become a MOC member to see the clickable quiz with answers on the back.

Become a MOC member to see the clickable quiz with answers on the back.

Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading
- THE ADDITION OF HYDROGEN CHLORIDE TO BUTADIENE
M. S. KHARASCH, J. KRITCHEVSKY, and F. R. MAYO
The Journal of Organic Chemistry 1937, 02 (5), 489-496
DOI: 10.1021/jo01228a010
An early paper by the esteemed chemist M. S. Kharasch on the addition of HCl to butadiene, seeing whether the ratio of 1,2 to 1,4 addition varied with different reaction conditions. - THE PEROXIDE EFFECT IN THE ADDITION OF REAGENTS TO UNSATURATED COMPOUNDS. XIII. THE ADDITION OF HYDROGEN BROMIDE TO BUTADIENE
M. S. KHARASCH, ELLY T. MARGOLIS, and FRANK R. MAYO
The Journal of Organic Chemistry 1936, 01 (4), 393-404
DOI: 10.1021/jo01233a008
Another early study by M. S. Kharasch in which he studies the addition of HBr to butadiene, in which he attempts to rigorously separate the two modes of addition – electrophilic vs. radical. - —Mobile-anion tautomerism. Part I. A preliminary study of the conditions of activation of the three-carbon system, and a discussion of the results in relation to the modes of addition to conjugated systems
Harold Burton and Christopher Kelk Ingold
J. Chem. Soc. 1928, 904-921
DOI: 10.1039/JR9280000904
A classic paper from a pioneer of physical organic chemistry, Prof. C. K. Ingold. He suggests the general idea that the 1,2 and 1,4 addition products are in equilibrium and that the 1,2-product can reverse reaction and form the 1,4 product. This is the basis of what we now call “kinetic” and “thermodynamic” control. - The modes of addition to conjugated unsaturated systems. Part IX. A discussion of mechanism and equilibrium, with a note on three-carbon prototropy
P. B. D. de la Mare, E. D. Hughes and C. K. Ingold
J. Chem. Soc. 1948, 17
DOI: 10.1039/JR9480000017
Here, C. K. Ingold has reviewed all work (including Kharasch’s) done up to that point in the study of electrophilic additions to conjugated systems. - Properties of conjugated compounds. Part XI. Addition of hydrogen bromide to βγ- and αδ-dimethylbutadiene
Ernest Harold Farmer and Frederick G. B. Marshall
J. Chem. Soc. 1931, 129
DOI: 10.1039/JR9310000129
When methyl groups are added to butadiene, the 1,2 and 1,4 addition ratios can change significantly! This is because the stability of the carbocation intermediates will change.
If the conversion of 1,2 product to 1,4 product is exothermic, how can it be favoured as the temperature increases? shouldn’t the equilibrium shift in the direction of the endothermic forward reaction by Le Chatelier’s principle?
I just want to thank you, thanks a lot !
You are welcome Khai, glad you’ve found it helpful.
Why does the more stable intermediate have a lower barrier to its transition state? Shouldn’t the opposite be true?
Hi,
Where can we find the solutions for the three examples toward the end?
Thanks!
J
Next post: https://www.masterorganicchemistry.com/2017/04/11/more-on-12-and-14-additions-to-dienes/
Difficult Concepts made so easy
Do you offer courses from scratch to proficient level
Do get back on my mail id
I am interested in a regular course that compels you to study
Am a tutor
Duration should be around 6 to 9 months
Is there ever a case where the 1,2 and 1,4 products are the same, regardless of temperature?
Sure. Cyclopentadiene. 1,2 addition of HBr and 1,4 addition of HBr gives you the same product: 3-bromocyclopentene