Free Radical Reactions
In Summary: Free Radicals
Last updated: May 7th, 2026 |
A Summary Of Topics For Free-Radical Reactions
So what have we learned in this series about free radicals? Lots of things. We’ve seen that they are neutral species with a half-filled valence shell that tend to be highly reactive with alkanes and double bonds. We’ve seen that they are stabilized by electron donors and delocalization. We’ve shown their mechanisms proceed through three distinct stages (initiation, propagation, termination) and that although reactions tend to be selective for formation of the more stable free radical, bromine radicals are far more selective than chlorine radicals in halogenation of alkanes. Finally we’ve also explored allylic (and benzylic) halogenation and seen examples of allylic rearrangement.
Table of Contents
- What Are Free Radicals?
- Homolytic Cleavage And “Single-Barbed” Curved Arrows
- Factors That Affect Free Radical Stability
- Factors That Destabilize Free Radicals
- A Handy Shortcut For Determining Radical Stability
- How Do Free Radical Reactions Work? Initiation, Propagation, Termination
- Free-Radical Halogenation Leads To Mixtures Of Isomers – But There Is Some Selectivity
- Bromination Is More Selective Than Chlorination
- Allylic Bromination, And Allylic Bromination With Allylic Rearrangement
- Free-Radical Addition Of Radicals To Alkenes
- Notes
- Quiz Yourself!
1. What Are Free Radicals?
Free radicals are chemical species that contain a singly occupied orbital. They are neutral and tend to be highly reactive.
One of the most common classes of free-radical reactions is free-radical substitution. Here is an example of the reaction between methane and Cl2:
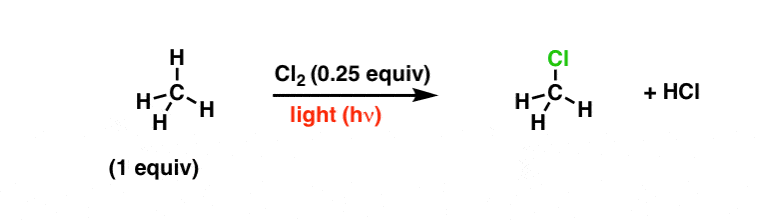
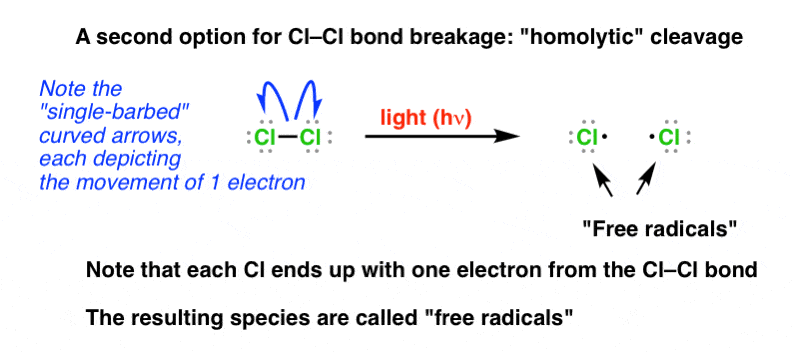
2. Homolytic Cleavage And “Single-Barbed” Curved Arrows
Unlike many of the reactions we have seen previously, free radical reactions do not involve the donation or acceptance of an electron lone pair and they are not ionic. Instead, free radical reactions operate through homolytic cleavage – that is, bonds break such that equal numbers of electrons are distributed to each atom. The curved arrow formalism is modified here: we draw curved arrows with a single barb, which shows the movement of a single electron:
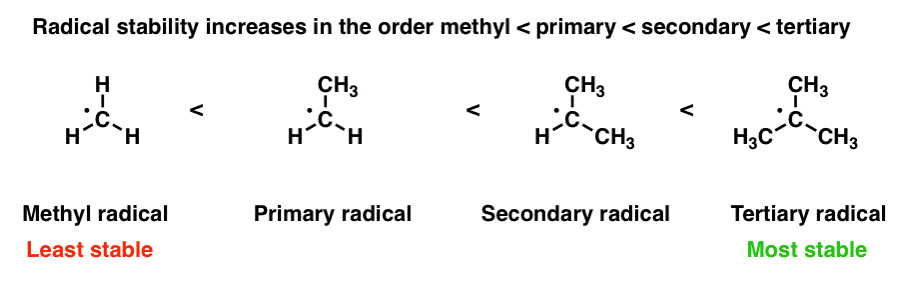
3. Factors That Affect Free Radical Stability
Like carbocations, free radicals are electron-deficient species. [Helpful to know: the factors which affect the stability of free radicals are the same which influence the stability of carbocations.] They can be stabilized through donation of electron density by neighbours; for this reason, radical stability increases in the order methyl < primary < secondary < tertiary . [Radicals are also stabilized by adjacent atoms with lone pairs, such as oxygen and nitrogen]. [See post: 3 Factors Which Stabilize Free Radicals]
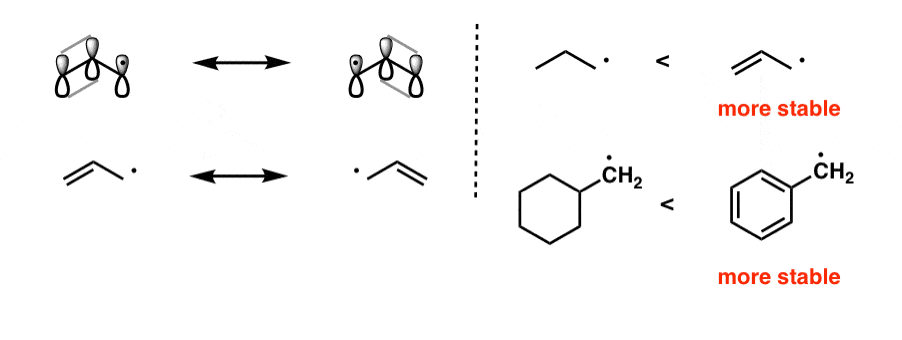
A second important factor which stabilizes free radicals is “delocalization” – that is, if the radical can be spread out over two or more carbons. A more familiar way of saying this is that free radicals are stabilized by resonance.
4. Factors That Destabilize Free Radicals
As electron-deficient species, it is also helpful to keep in mind some of the factors which destabilize free radicals. There are several important trends to keep in mind [See post: Which Factors Destabilize Free Radicals?]
- Free radicals decrease in stability as the % of s-character in the orbital increases [i.e. as the half-empty orbital becomes closer to the nucleus]. For that reason, free radical stability decreases as the atom goes from alkyl to alkenyl to alkynyl.

- Across a row of the periodic table, free radicals decrease in stability as the electronegativity increases

- Free radicals increase in stability going down a column of the periodic table, F• < Cl• < Br• < I• since the electron-deficient orbital is spread out over a greater volume.
- Free radicals adjacent to an electron-withdrawing group are less stable, since in effect, electron-density is being taken away from what is already an electron deficient species. [Watch out, however – this only applies to electron withdrawing groups that cannot donate a pair of electrons, like CF3 or CN.]
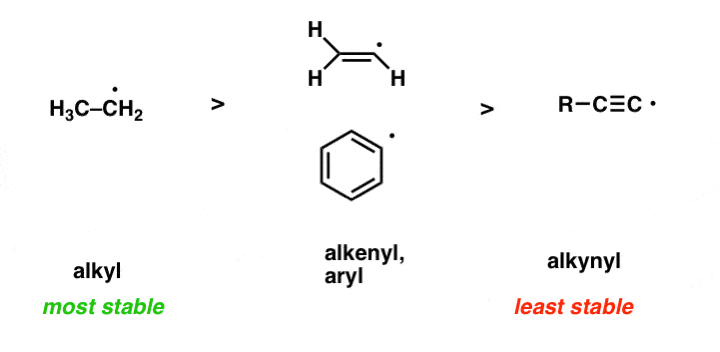
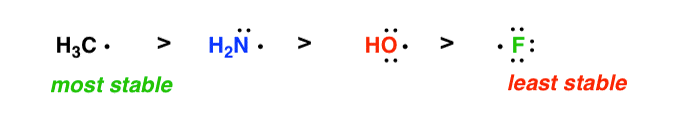
5. A Handy Shortcut For Determining Radical Stability
Bond dissociation energies [BDE’s] measure the energy required for homolytic bond cleavage, and by proxy we can interpret C–H bond strengths as a good measure of carbon free-radical stability. By examining trends in X-H bond strengths (where X is various atoms) we can discern trends in radical stability. [See post: Bond Strengths and Radical Stability]
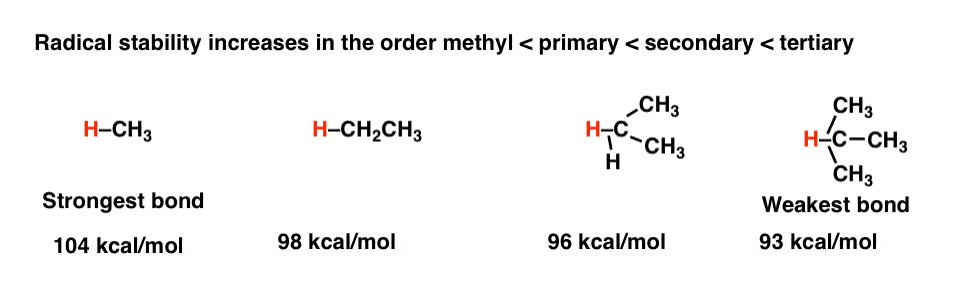
This is also reflected in the strengths of “allylic” C-H bonds, which are weaker than normal due to the fact that a resonance-stabilized free-radical is produced. For example, the C-H bond strength of the methyl group of propane is about 98 kcal/mol, whereas that of the methyl group of 1-propene is 85 kcal/mol. For a complete list of factors, refer to the original post.
6. How Do Free Radical Reactions Work? Initiation, Propagation, Termination
One “sure fire” way students find themselves identifying free-radical reactions is to look for the presence of “heat” [ Δ] or “light” [hν ]. What does heat or light have to do with free-radical reactions? Well, it facilitates the first important step in any free radical reaction: initiation. This is the homolytic fragmentation of a weak bond to form two new free radicals. [See post: Free Radical Reactions – Why is “Heat” or “Light” Required?]
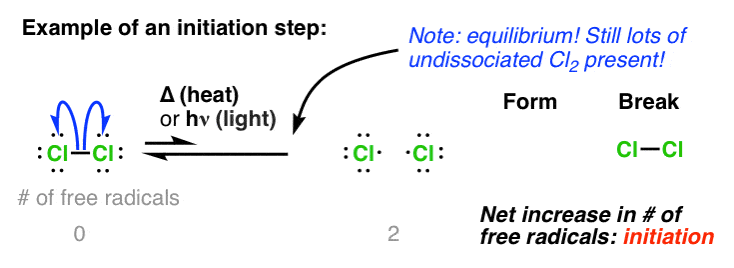
It’s important to note here that “initiation” is not a reaction that goes to completion. At any given point, only a small concentration of the starting material is converted to free radicals. However, since the reaction is a chain-reaction, this is generally sufficient.
The second step in a free-radical process is at least one (but often two) “propagation” steps. For the free-radical halogenation of alkanes, the first propagation step is removal of hydrogen by a halogen radical, giving a new carbon radical. Note here that the number of free radicals remains constant (one on both sides of the equation).
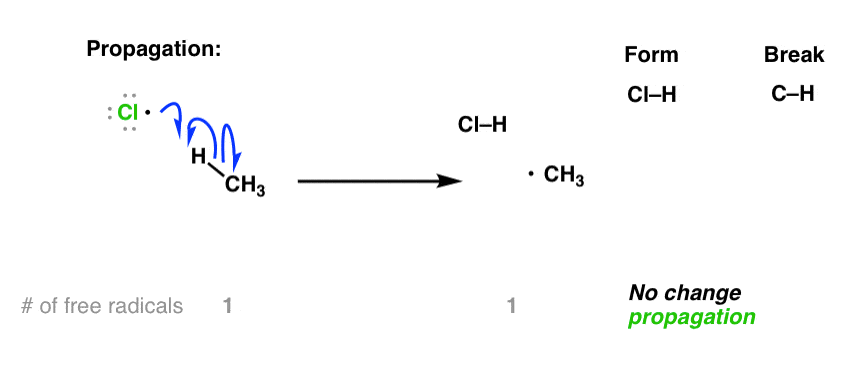
This is followed by a second propagation step. While it may be tempting for many new students to form CH3-Cl through combination of CH3• with a chlorine radical, that’s not what happens next [that would technically be a “termination” step, see below]. What happens next is the reaction of alkyl radical with one of the starting molecules of Cl2, forming a new C–Cl bond and a new Cl• radical which continues the chain reaction. Again, note: no net increase in the number of free radicals here:
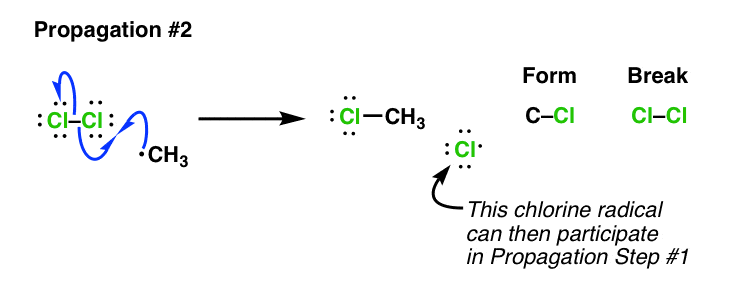
At any given point the concentration of free radicals in solution is quite low. However, after most of the Cl2 is consumed, free-radical concentrations will start to increase until the rate of radical-radical combination reactions is significant. This is called “termination” since it results in a net destruction of free radicals. There are many potential ways this can occur – here is one example:
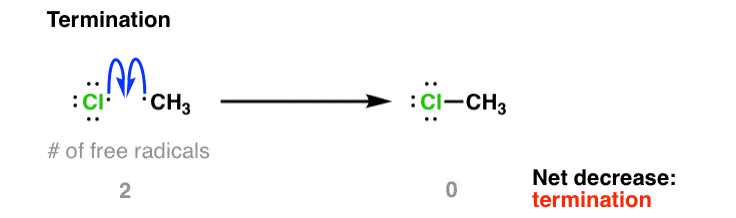
The bottom line is that by counting the number of radicals created or destroyed in each step you can determine if the step is initiation, propagation, or termination.
[See post: Initiation, Propagation, Termination]
7. Free-Radical Halogenation Leads To Mixtures Of Isomers – But There Is Some Selectivity
Methane and ethanes are simple examples. Replacement of any one of the 4 hydrogens of methane by chlorine will result in the same product. However when we are dealing with a more complex alkane, like propane, butane, or 2,3-dimethyl hexane, we must be alert for the possibility that a variety of compounds will form. It’s an important skill to be able to recognize all the possible compounds that could result from, say, the mono-chlorination of an alkane like 2-methylpentane [there are 5]. These types of questions are best approached systematically: start with replacement of hydrogen by chlorine on one side of the molecule, and gradually work to the other side – it can help to double check that there are no duplicates by trying to name them (IUPAC). [See post: Isomers From Free Radical Reactions]
That being said, isomers will not be formed in equal quantities. There are two factors to consider. The first is the fact that not all C-H bonds are of equal strength; as we mentioned above, tertiary C-H bonds are easier to break than secondary C-H bonds, which are easier to break than primary C-H bonds. So there will be an inherent difference in reactivity between these bonds simply by the fact that it requires less activation energy to break a weaker bond. A second factor is statistics. In propane, for example, there are six primary C-H bonds and two secondary C-H bonds. Based on statistics alone, we would expect mono-chlorination propane to yield a 3:1 ratio of 1-chloropropane to 2-chloropropane.
What’s interesting is that the actual ratio of 2-chloropropane to 1-chloropropane is 45:55 !
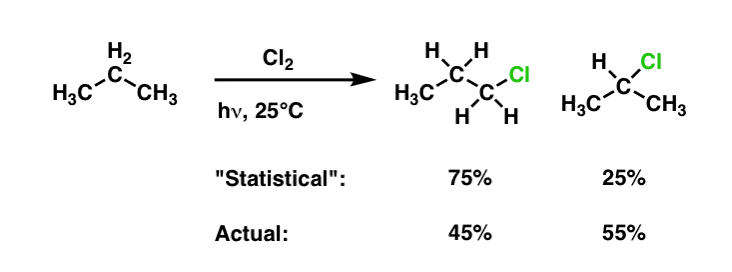
It’s possible to use this information to quantify the reactivity of each type of hydrogen in a molecule. Using the method from this post, we can determine that the secondary C-H is 3.66 times more reactive than the primary C–H under these conditions. [See Post: Selectivity In Free Radical Reactions]
8. Bromination Is More Selective Than Chlorination
Interestingly, bromination of alkanes is even more selective than chlorination for tertiary and secondary C–H bonds. For example in the bromination of propane at 25°C, the reaction is selective for secondary C–H over primary C–H by a factor of 97 to 1 .
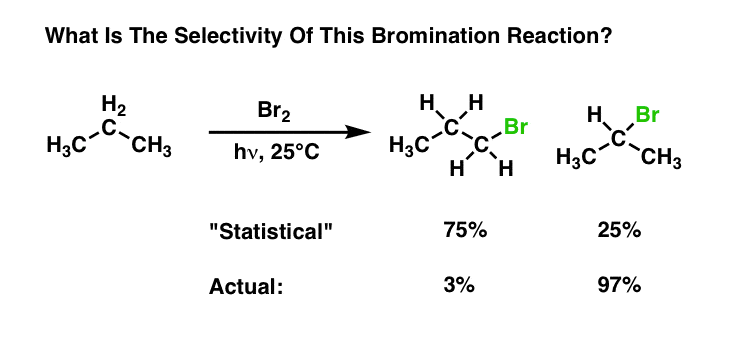
Why the huge difference? The most rigorous way to look at it is to apply Hammond’s Postulate: chlorination is exothermic, bromination is endothermic; therefore, chlorination will have a “reactant-like” transition state and bromination will have a “product-like” transition state. In this case the difference in energies between products is greater than the difference in energies between reactants ; and since selectivity is determined by differences in energies between transition states, therefore, bromination will be more selective than chlorination. If this sounds complex, the full post is here. Alternatively, a simple analogy I use to explain this concept is here. [See Post: Selectivity In Free Radical Reactions – Chlorine vs. Bromine]
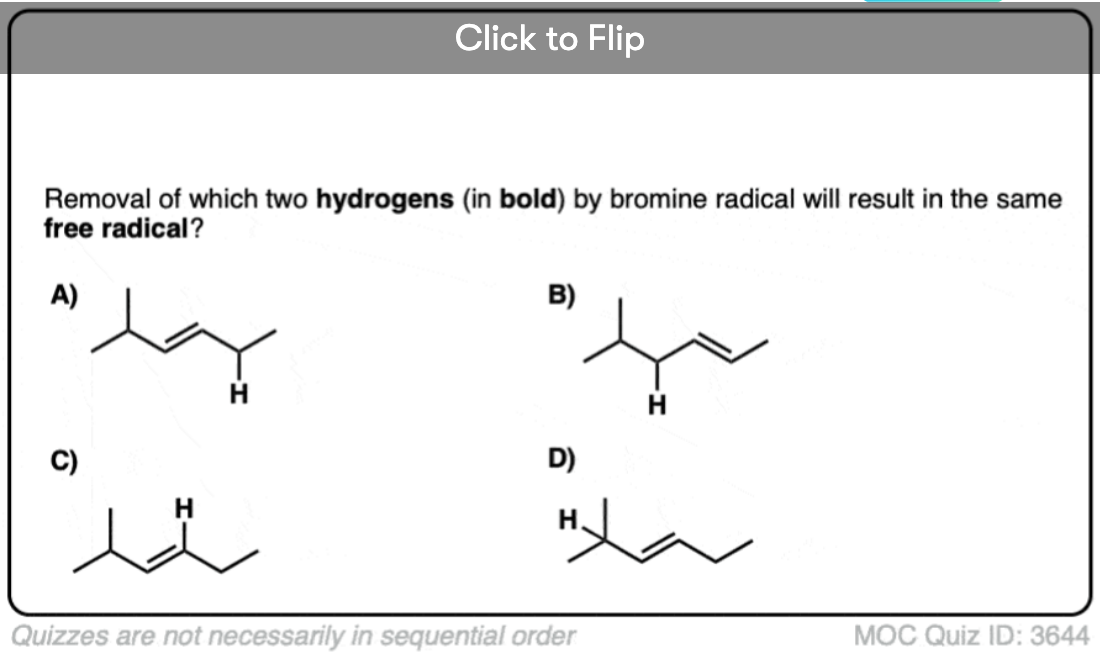
9. Allylic Bromination, And Allylic Bromination With Allylic Rearrangement
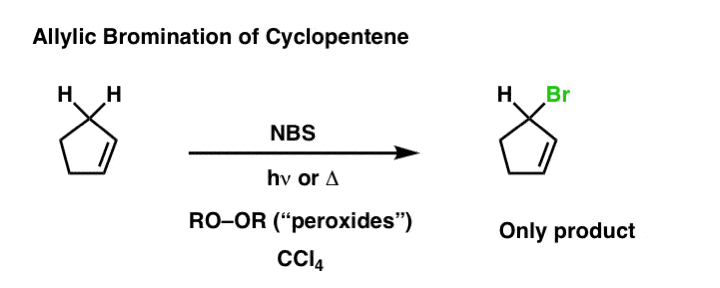
Weak C–H bonds (like tertiary C–H bonds, for instance) are very easily halogenated. Allylic and benzylic C–H bonds are particularly weak (about 86 kcal/mol) which makes them excellent substrates for bromination reactions. Molecular bromine (Br2) can be useful for benzylic bromination, but attempts to use this reagent with alkenes like, say, propene, would result in formation of a dibromide.
The way around this is to use the useful source of bromonium ion, N-bromo succinimide (NBS), which serves as a dilute source of Br2 . This allows replacement of the allylic C–H bond with Br. This process is called “allylic bromination“.
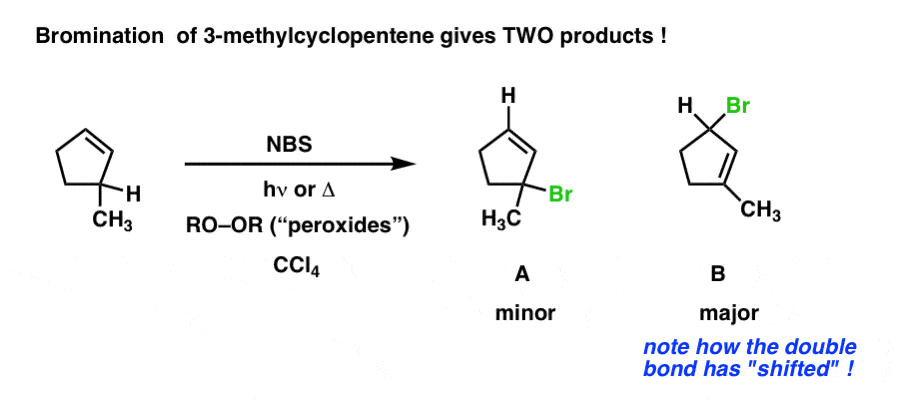
Interestingly, changing the substrate just a little bit can result in some surprising results. The example below differs from the example above by only a single methyl group. However, this can lead to two potential products – the “normal” product of allylic bromination (A) and a closely related product, allylic bromination with allylic rearrangement (B). One factor which can dictate which final product is formed is determining which alkene is more substituted.
10. Free-Radical Addition Of Radicals To Alkenes
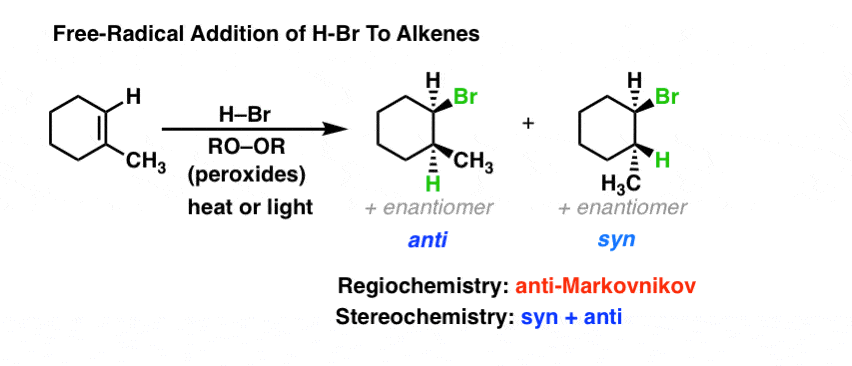
Finally, it’s important to note that H–Br can add to alkenes in a completely different fashion depending whether or not “peroxides” are present. In the absence of peroxides, H-Br adds through a “normal” electrophilic addition [post here – not radical related]. However, if peroxides are added and the mixture is heated, it leads to homolytic cleavage of the peroxide, forming new free radicals (initiation). Peroxide can then remove a hydrogen from H-Br forming the Br• radical, and then Br• can add to alkenes. What’s important to note is that Br• adds to alkenes in a such a way that we end up with the most stable radical intermediate, which then removes hydrogen from H–Br, regenerating bromine radical. The net result is “anti-Markovnikov” addition of H-Br across a double bond, the opposite of what is observed in the absence of peroxides. [full mechanism in this post]
That does it for free radical reactions (for now).
In the next series we’ll start putting a lot of our Org 1 reactions together… and finally delve into one of my favourite topics – synthesis.
Notes
Note 1. Related to the addition of H-Br to alkenes, is an interesting process called “free radical polymerization”.
If certain alkenes are heated in the presence of a radical initator (let’s say “peroxides” as a typical example), the peroxy radical can add to the alkene, forming a new free radical on the most substituted position… and the resulting free radical can add to another alkene…. generating another free radical…. which adds to yet another alkene! – and so on.
The process can repeat thousands of times before termination occurs. Some of the most common plastics of daily life can be made this way – such as polyethylene, polypropylene, and polystyrene.
Quiz Yourself!

Become a MOC member to see the clickable quiz with answers on the back.
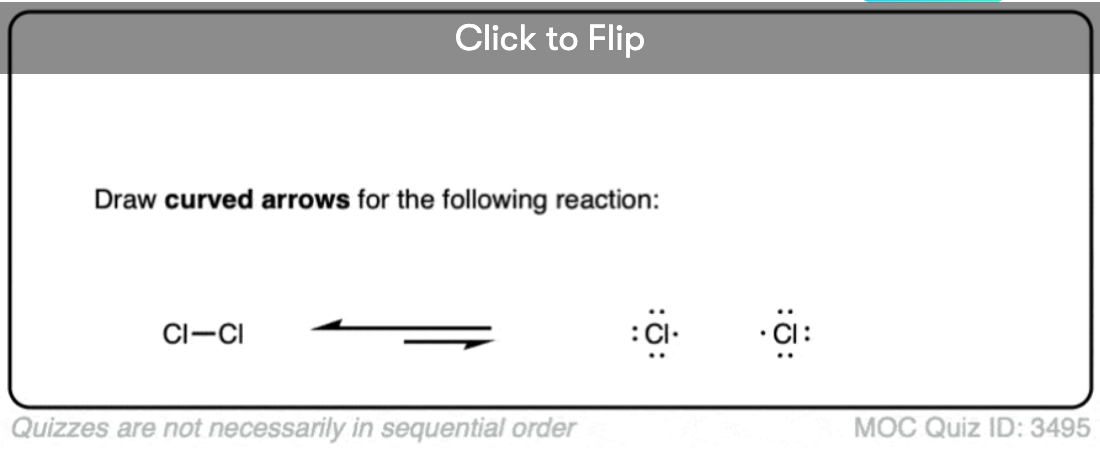
Become a MOC member to see the clickable quiz with answers on the back.
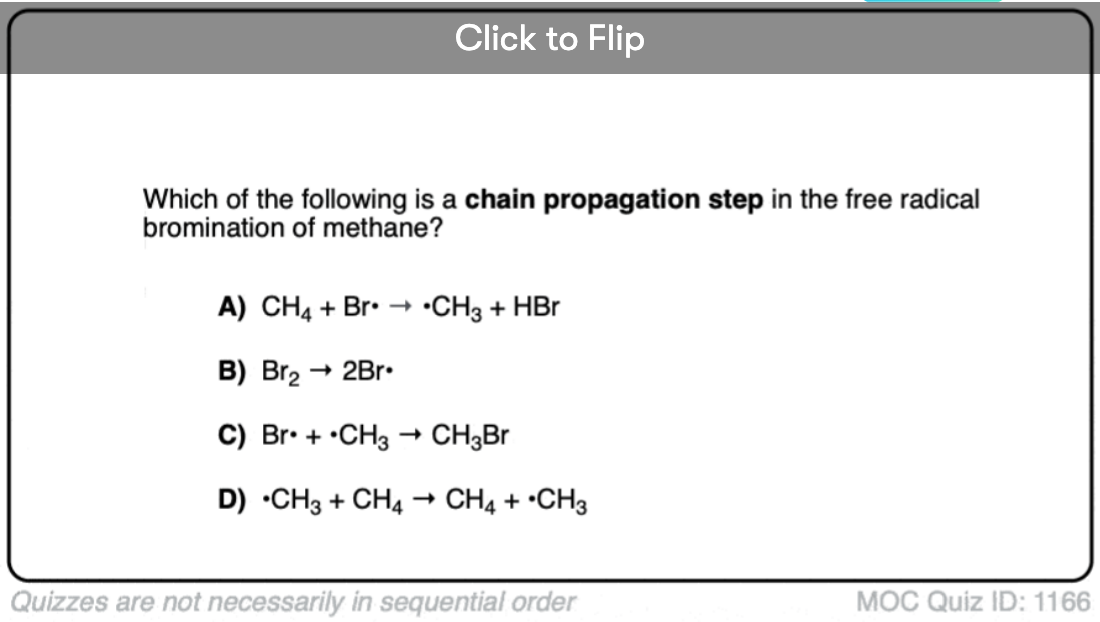
Become a MOC member to see the clickable quiz with answers on the back.
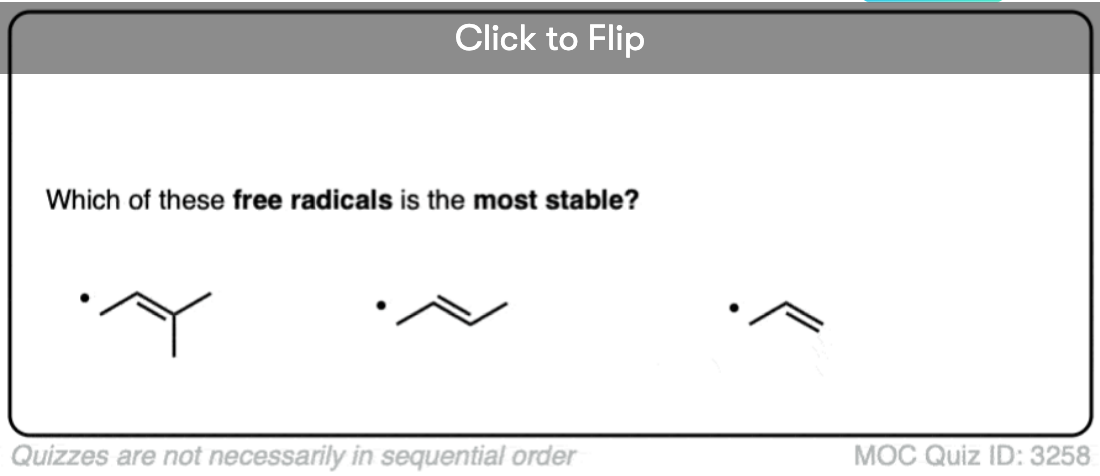
Become a MOC member to see the clickable quiz with answers on the back.

Become a MOC member to see the clickable quiz with answers on the back.
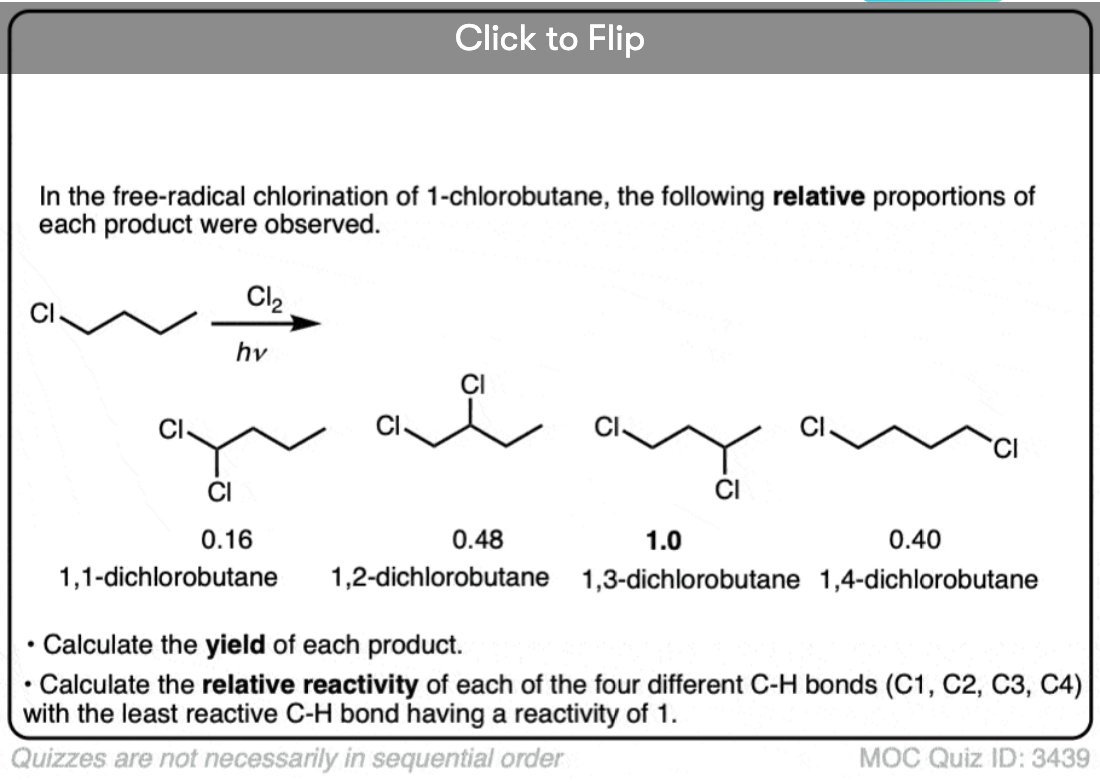
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Under point 5 “What’s interesting is that the actual ratio of 2-chloropropane to 1-chloropropane is 45:55!”
Should be 1-chloropropane to 2-chloropropane, according to the other post about selectivity.
Thanks to this post series, I don’t think I’ll ever forget the free radical mechanism now.
Fixed – thank you very much
Hi James, I was wondering why free radicals of bromine or chlorine do not cause polymerization like peroxides?
There’s nothing to say that they *can’t*, specifically, although with alkenes, the precursors of bromine and chlorine radicals also tend to add as electrophiles to double bonds (e.g. forming bromonium and chloronium ions) which would not be good for polymerization. Peroxides don’t tend to have these cationic pathways.
Under point 3, factors that destabilize the free radical you have mentioned that free radicals next to electron withdrawing groups are less stable right? There you have also mentioned than cyanide CN cannot donate an electron. Could you explain why can’t it? I dont see it donating electrons through its lone pair as in amines or hydroxides but can’t it do so through conjugation of its pi electrons as , for instance, phenyl does?
(p.s The Font is super satisfying)
Why does the radicals favor the C-H bond over the C-Cl bond that formed recently? Assuming you have another solution where only you have CCl4 compound, would radicals such as hydroxel radicals be able to break C-Cl bonds or not? if so, which C-halogen are more sensitive to hydroxel radical attacks C-Cl, C-Br or C-I??
The reaction will favor breaking of C-H over C-Cl bonds because the resulting H-Cl bond (103 kcal/mol) is stronger than the resulting O-Cl bond (65 kcal/mol) . The O-Cl bond is quite weak due to repulsion between the lone pairs. The driving force for a radical breaking C-Cl is not strong enough.
Dear sir,what 1 degree ,2 degree free redical means?
1° is a primary free radial where the carbon bearing the radical is attached to one other carbon. 2° is a secondary free radical where the carbon bearing the radical is attached to two other carbons.
Dear Mr. Ashenhurst,
I do not understand why the radicals are neutral. Why are they not positive?
Is that not right that they want to fill their orbital?
Can you explain it me, please?
Thanks for your help.
Yours faithfully,
Marie B.
It’s worth recalling that atomic carbon, C , has four valence electrons, four empty places in its octet, and is neutral.
Atomic fluorine, F• , has seven electrons, one electron away from a full octet, and is also neutral.
Just because a species is reactive and has a high electron affinity does not mean that it has to be charged.
For proof, go to the formal charge formula for the methyl radical, H3C• . If carbon has 4 electrons to itself, it is neutral. Here it has one electron to itself (the radical) and has a 50% share in the six bonding electrons of the C-H bonds, (3) giving a total of 4. If it loses the electron it becomes the methyl cation, CH3(+). If it gains an electron it becomes the methyl anion, CH3(-).
So, what is more important in product formation — intermediate radical stability or product stability (alkene substitution)?
I don’t know how to answer your question directly. If you are asking, “what determines what product will be formed”, the answer lies in comparing the activation energies for the various competing rate-determining steps.