Dienes and MO Theory
What To Expect In Organic Chemistry 2
Last updated: November 1st, 2022 |
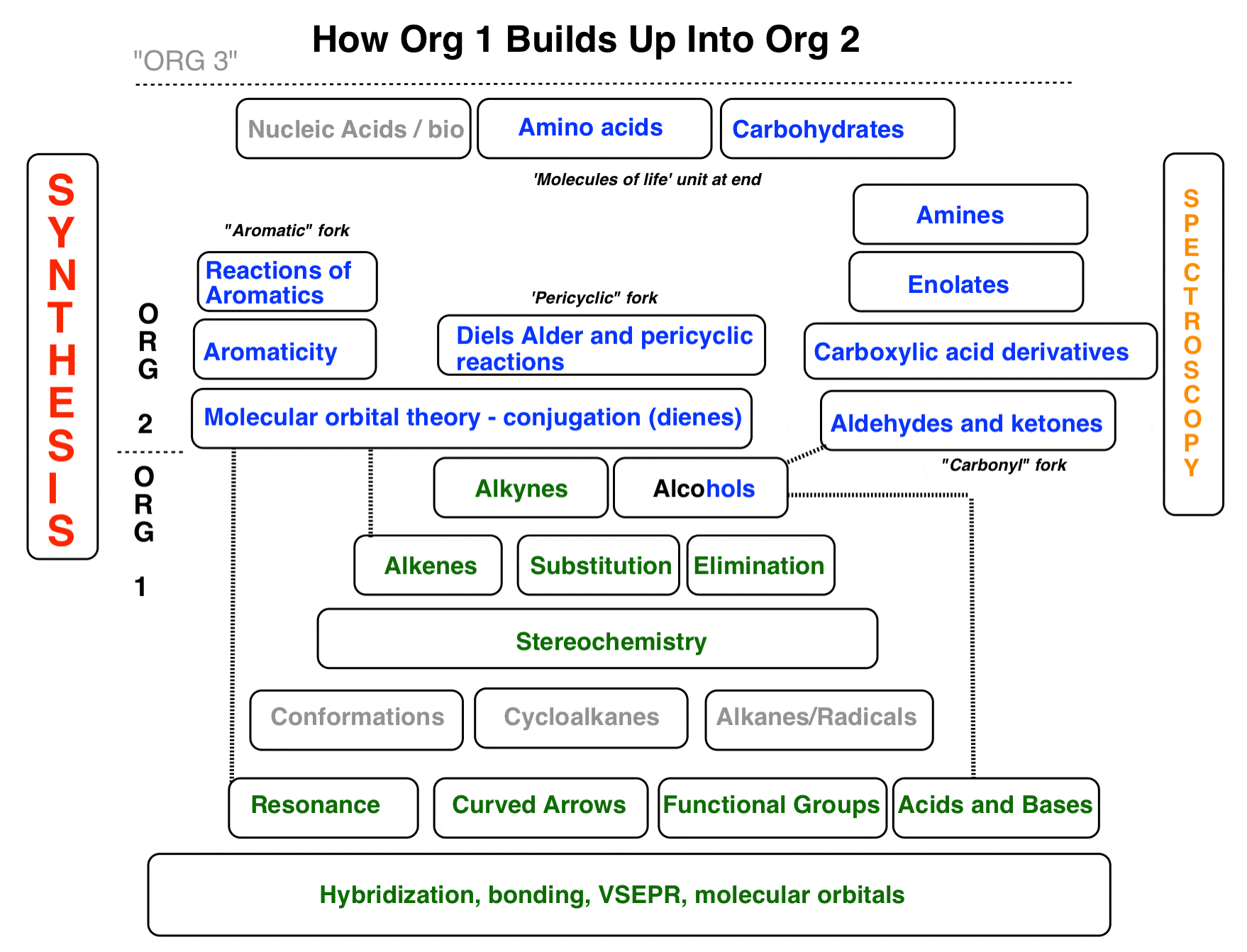
Organic Chemistry 2 Is Largely About “Resonance”
A large part of organic chemistry 1 is devoted to laying the foundations: introducing structural concepts such as bonding, geometry, stereochemistry, conformations, resonance, and steric effects, while introducing concepts in chemical reactivity such as nucleophilicity, electrophilicity, acidity, basicity and so on.
While there are several important new concepts introduced in Org 2 (especially in the context of pi-bonding) you will find that it largely builds on the foundations of Org 1 and assumes that you fully understand these concepts. The focus in Org 2 will be much more on reactions.
If I had to name an overall theme for Org 2, it would be : “the chemistry of pi bonds.” Understanding resonance will be very important!
Here is a brief introduction to some of the highlights of Org 2. By the end of the course, the answers to the following questions hopefully won’t seem as mysterious.
Table of Contents
- Conjugation, And Why It Matters
- Thermodynamic And Kinetic Control
- Cycloaddition Reactions
- Aromaticity
- Carbonyl Chemistry
- Biomolecules
- Summary: What To Expect In Organic Chemistry 2
1. Conjugation, And Why It Matters
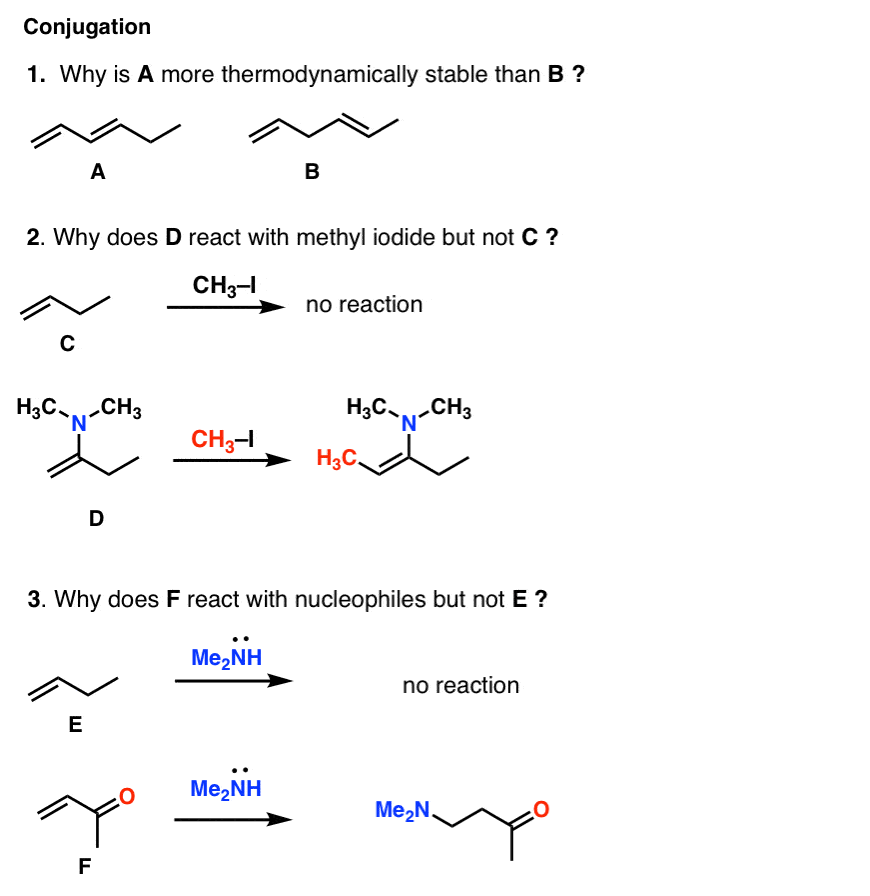
Why is it that diene A is more stable than diene B? How is it that replacing a single substituent on an alkene can drastically affect the reactivity? For example, why is it that alkene C does not react with electrophiles such as alkyl halides, but alkene
D does. And alkene E does not react with nucleophiles, but alkene F does. How do we explain this?
2. Thermodynamic And Kinetic Control
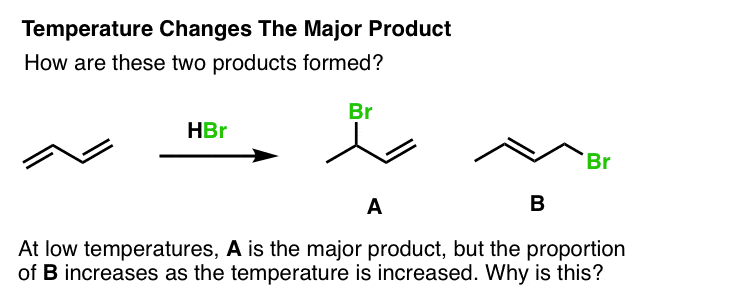
In the addition of HBr to an isolated alkene such as, say, 1-butene, there is only one product possible (not counting stereoisomers). However, if we add HBr to butadiene, we can get two products, A and B. At low temperature, we obtain A as the dominant product, but at high temperature, B is dominant. Why is this?
3. Cycloaddition Reactions
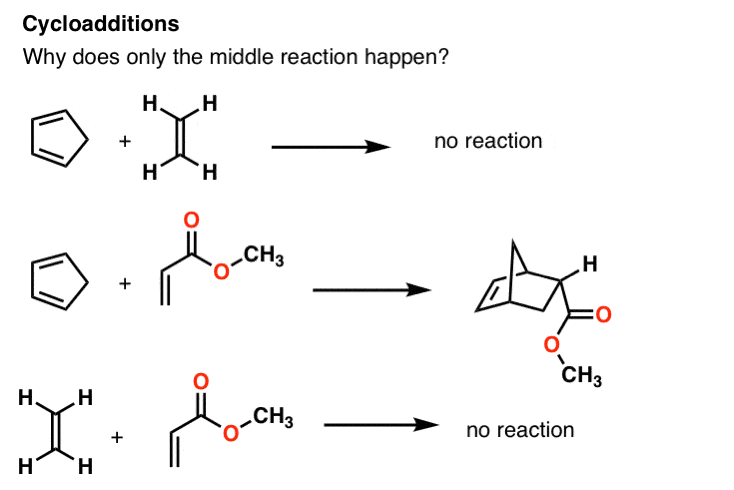
When the diene (cyclopentadiene) is added to ethene, no reaction occurs. But when we use the middle alkene instead, there is a rapid reaction to provide a new cyclic compound. What is different about this alkene that allows this reaction to occur? And why will this alkene react readily with cyclopentadiene, but when it is added to ethene, nothing happens?
4. Aromaticity
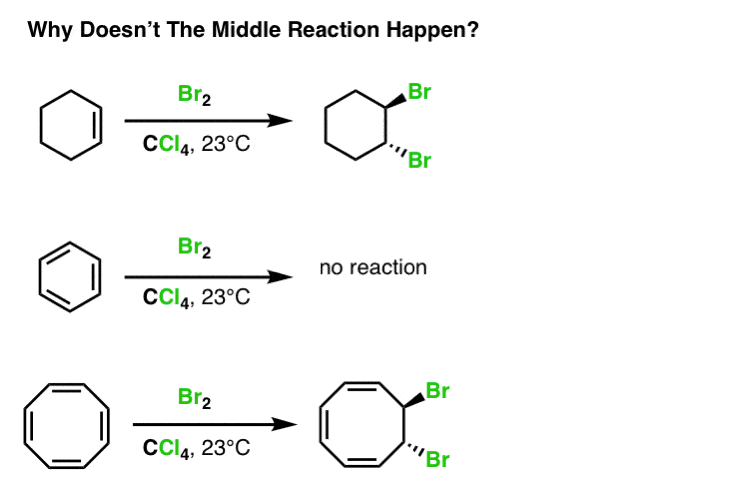
Normal alkenes (like cyclohexene) give trans dibromides when treated with Br2. But the cyclic triene in the middle (benzene) doesn’t react with bromine at all (unless you add a catalyst, and even then you get a different product!) Why the difference in reactivity? And why is this behavior similar for molecules like furan, pyrrole, and pyridine, but a molecule like cyclooctatetraene (bottom) will also add Br2?
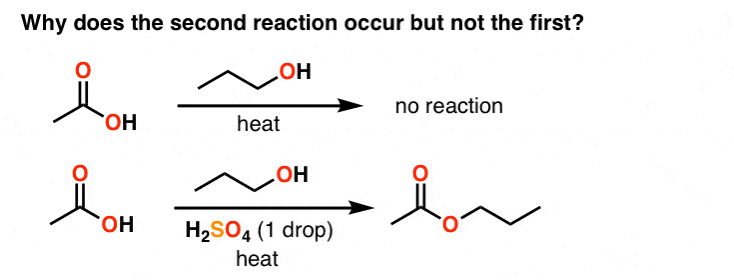
5. Carbonyl Chemistry
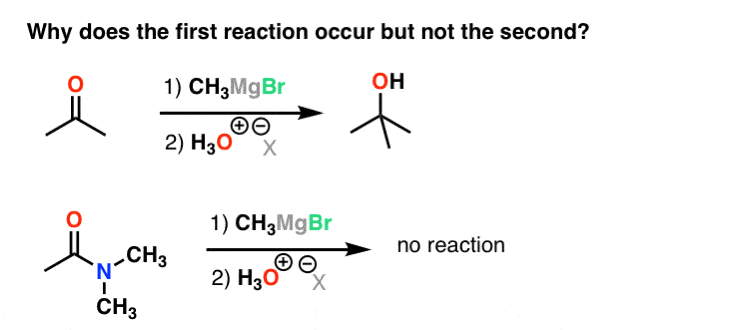
Grignard reagents react readily with aldehydes and ketones, but not amides. What causes the difference in reactivity?
When an alcohol is added to a carboxylic acid, the flask has less mojo than a Shaker meetinghouse. But when just a drop of acid is added, they readily combine to form an ester. Why is this?
6. Biomolecules
Your body is made up of proteins, sugars, and fats. What do these molecules look like? What are their properties? How can we make them in the laboratory, and potentially modify their structures?
7. Summary: What To Expect In Organic Chemistry 2
It’s vitally important to: make sure you have a firm handle on the key concepts and reactions from organic 1, because it will be assumed that you know it all.
The biggest challenge will be: getting a handle on the large number of reactions to learn, especially in carbonyl chemistry.
It will make your life easier if… you look for underlying trends and themes and see that underneath what appears to be tremendous variety, there are really only a handful of mechanisms.
The most important skill the course will provide: being able to design and plan multiple-step syntheses of relatively complicated molecules. Furthermore, the course will give you a much greater understanding of the chemistry of biologically important molecules such as amino acids, lipids, and sugars. It will be a lot of work, but you’ll see the world around you in a different way.
Edit – fixed some typos. Thanks to Prasanna for pointing them out.
P.S. Where does “Org 2” end and “Org 3” begin? I’d say it’s when we start having to deal with multiple functional groups in the same molecule that can potentially react. From Org 3 onwards, we really start to focus on the issue of “selectivity” : selectivity between functional groups (“chemoselectivity”), selectivity for synthesis of various stereoisomers (“stereoselective synthesis”), and designing the synthesis of ever more complex molecules from an ever-growing arsenal of reactions.
Thank u sir, am looking forward to see more of good work
Dear James,
Hello. Perhaps it might be a good idea if we add a hyperlink of this 2011 post “What to Expect in Organic Chemistry 2” https://www.masterorganicchemistry.com/2011/01/19/what-to-expect-in-organic-chemistry-2/
to the post “How Concepts Build Up in Org 1, ‘The Pyramid'” back in 2014.
https://www.masterorganicchemistry.com/2014/09/05/how-concepts-build-up-in-org-1/
And vice versa, with the other page linking to the other one. Just a thought, it’s entirely up to you.
Thank you as usual! Really great site. I’m just learning organic chemistry for fun.
Cheers,
Romeo
Thank you, that’s a great idea.
best way to learn o.c
Thanks James Such a wonderful website for easy understanding of organic chemistry.
James, thanks, thanks, thanks SO much for the wonderful website.
Thanks SO much for making OChem. way easier to understand.
Thanks! Glad you find the website helpful! Tell your friends!!!
Hey James, im taking Organic 2 next semester and am looking to get a head start. I used your website alot last semester and recieved an A in Organic 1. (Thank you). I was just wondering if the Organic 2 section on your blog covers the entire, or most of, a normal Organic 2 course, because the Organic 1 section seems a lot more full.
Organic Two will be difficult for me, given that I hate pi bonds…but I got a good grade in the previous class. Which would you say is harder, Orgo 1 or 2?
Thanks!
Hard to say. Org 1 is mostly about learning concepts and then applying them. Org 2 involves learning a lot of different reactions. It really depends on how the course is taught to you.
How about drawing sterioisomers, Fischer projections, enantiomer, diasteromer, physical properties of different sterioisomers, etc
Stereochemistry isn’t nearly as important in Org 2. You’re expected to *know* it, but it isn’t tested as much.
How important is stereochemistry in orgo 2. Do they expect you to name R and S ?
yes, they do. In orgo 2 right now.
Thanks James – I’ll keep you updated on how its going.