A Primer On Organic Reactions
3 Factors That Stabilize Carbocations
Last updated: May 28th, 2026 |
Carbocations: Properties, Formation, and Stability
- Carbocations are electron-deficient species with an empty p-orbital
- Lacking a full octet and bearing a positive charge, they tend to be fairly high-energy (i.e. unstable) species and are often encountered as transient intermediates in many chemical reactions.
- Three main factors increase the stability of carbocations:
- Increasing the number of adjacent carbon atoms: methyl (least stable carbocation) < primary < secondary < tertiary (most stable carbocation)
- Adjacent pi bonds that allow the carbocation p-orbital to be part of a conjugated pi-system system (“delocalization through resonance“)
- Adjacent atoms with lone pairs that can provide the carbon with a full octet.
- Adjacent electron withdrawing groups that are unable to donate a lone pair (e.g. CF3, NO2 ) greatly decrease the stability of carbocations.
- Additionally, carbocations decrease in stability in the order alkyl (most stable) > alkenyl > alkynyl (least stable)
- A few additional factors that influence carbocation stability include aromaticity / antiaromaticity, planarity (bridgehead carbocations are unstable) and special cases involving small rings.
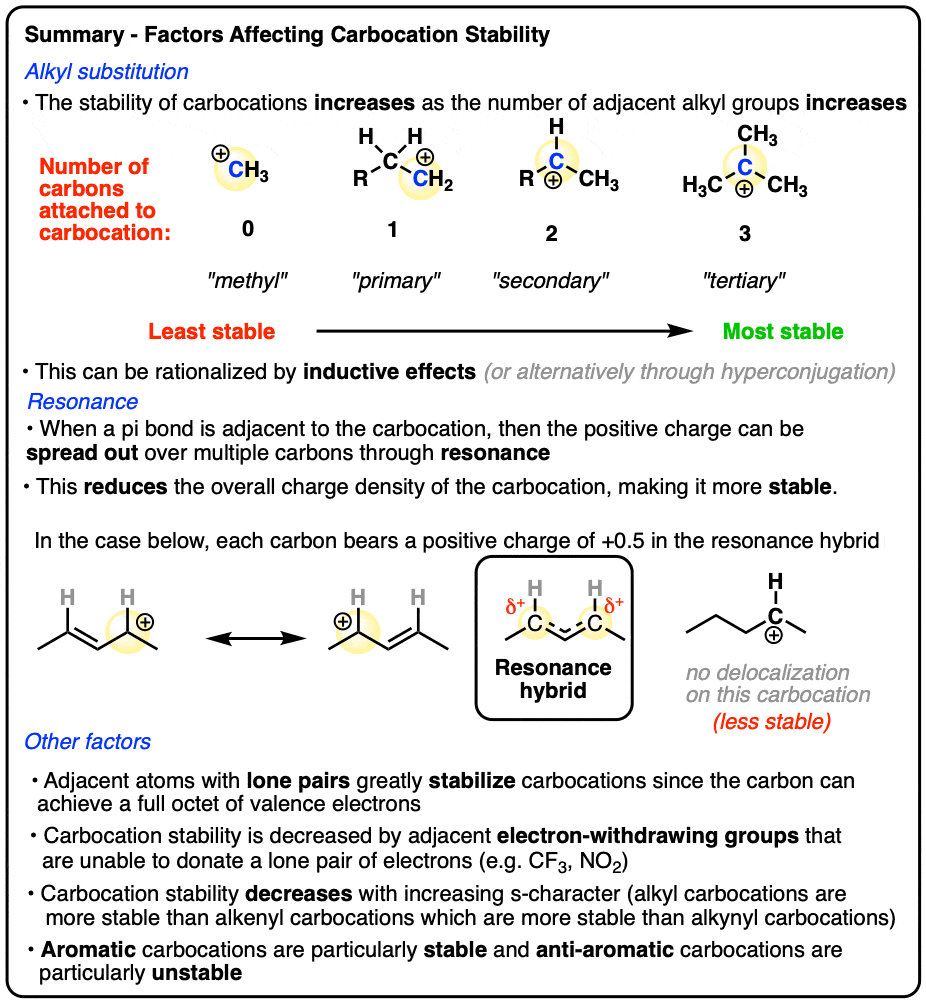
Table of Contents
- What Is A Carbocation?
- Formation of Carbocations
- Factors That Stabilize Carbocations – Substitution
- Factors That Stabilize Carbocations – Resonance
- Applying Carbocation Stability To Understand Reactions
- Stabilization By Adjacent Lone Pairs
- What’s More Important – Resonance Or Substitution?
- Inductive Effects
- Some Special Cases
- Summary
- Notes
- Quiz Yourself!
- (Advanced) References and Further Reading
1. What is A Carbocation?
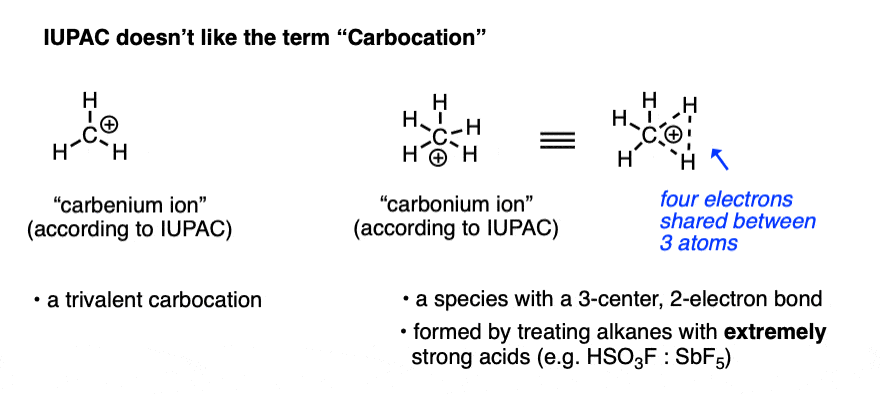
A positively charged carbon atom bearing three covalent bonds and an empty orbital is called a carbocation (or more officially, a “carbenium” ion, although for our purposes we’re going to use “carbocation” [Note 1 ])
Other than the charge of +1 on the central carbon, the structure and properties of the vast majority [Note 2] of carbocations closely resemble those of neutral boron compounds.

Both carbocations and neutral boron compounds generally have
- an sp2-hybridized central atom
- with 6 valence electrons,
- an empty p-orbital,
- trigonal planar geometry
- and bond angles of 120°.
[How do we know this? X-ray crystallography Note 3]
Like boron compounds, carbocations are electron-deficient Lewis acids that will readily combine with Lewis bases, resulting in a tetrahedral, sp3-hybridized atom with a full octet of electrons.
See if you can draw the curved arrow for the reaction below.
 Click to Flip
Click to Flip
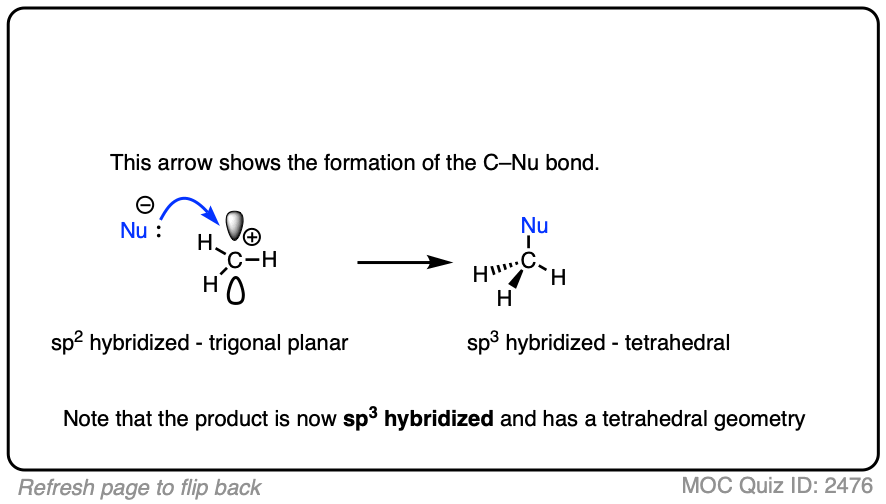
Due to their trigonal planar geometry, carbocations can undergo attack from either face of the empty p-orbital.
Be alert for situations where this can result in a pair of stereoisomers.
 Click to Flip
Click to Flip
2. Formation of Carbocations
Carbocations are important intermediates in many reactions. (An intermediate, as opposed to a transition state, is a potentially isolable species in a reaction and occupies a potential energy minimum in a reaction coordinate diagram. Transition states have partial bonds, extremely short lifetimes and cannot be isolated).
Lacking a full octet of electrons around carbon and bearing a positive charge, carbocations are higher in energy and more unstable than neutral carbon compounds.
Some prominent reactions that involve carbocation intermediates are the addition of hydrogen halides (e.g. HBr, HCl, and HI) to alkenes and unimolecular substitution (SN1) and elimination (E1) reactions of alkyl halides.
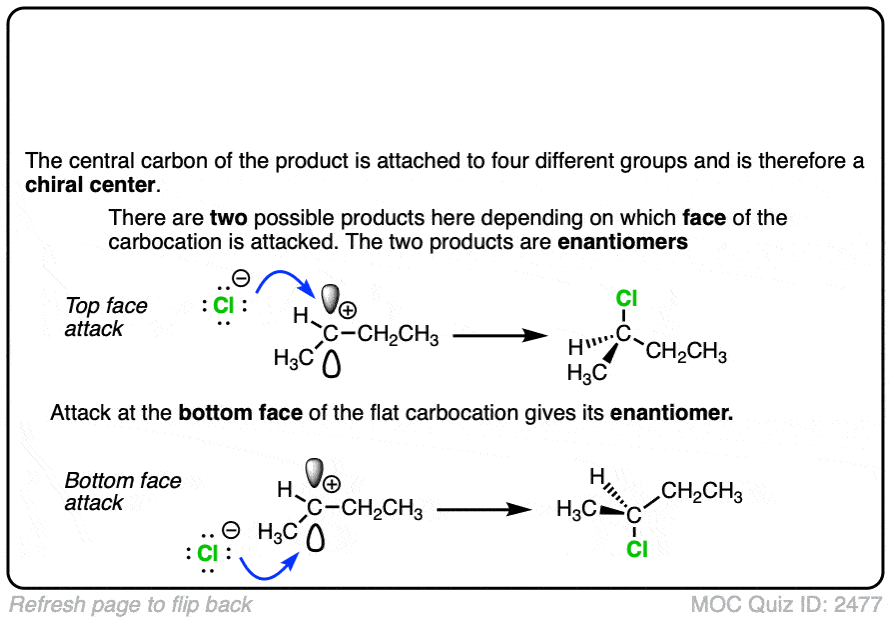
In the SN1/E1 reactions, the first step is loss of a good leaving group (generally, a very weak base – See “What Makes A Good Leaving Group“) to give a carbocation. This is usually done in a highly polar solvent such as H2O or acetic acid which can help to stabilize the charged carbocation intermediate.
See if you can draw the curved arrow for formation of the carbocation from this tertiary alkyl halide, along with the transition state:
 Click to Flip
Click to Flip
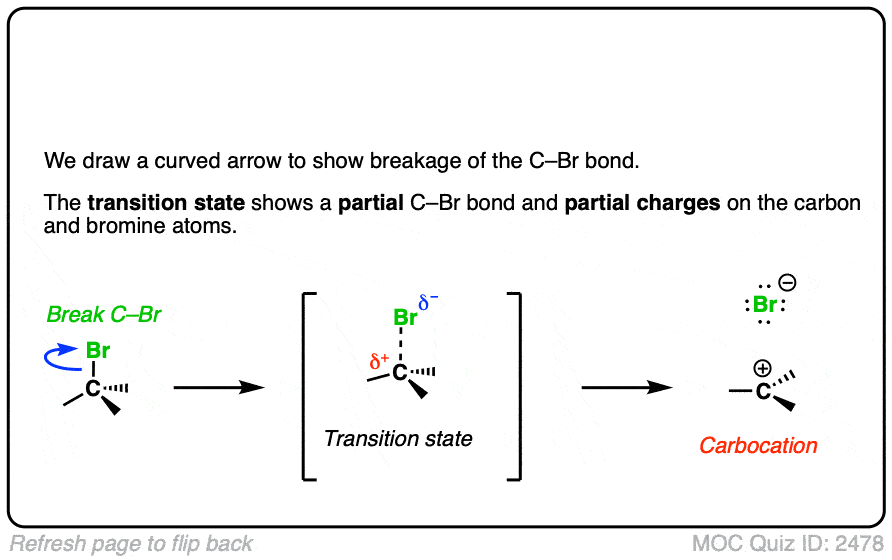
In additions of hydrohalic acids to alkenes, the first step is attack of H+ by the pair of electrons in the C-C pi bond to give a carbocation intermediate.
See if you can draw the curved-arrow pushing mechanism and the transition state for this reaction:
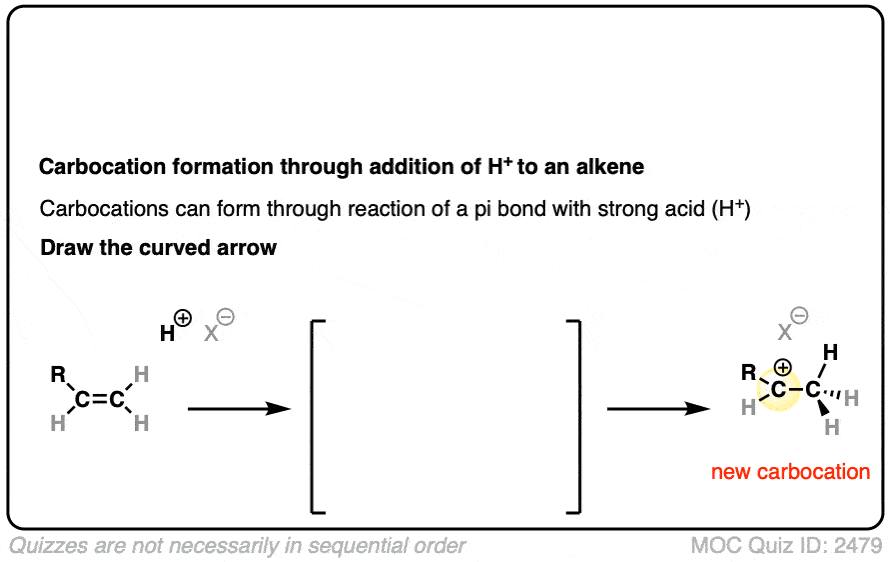 Click to Flip
Click to Flip
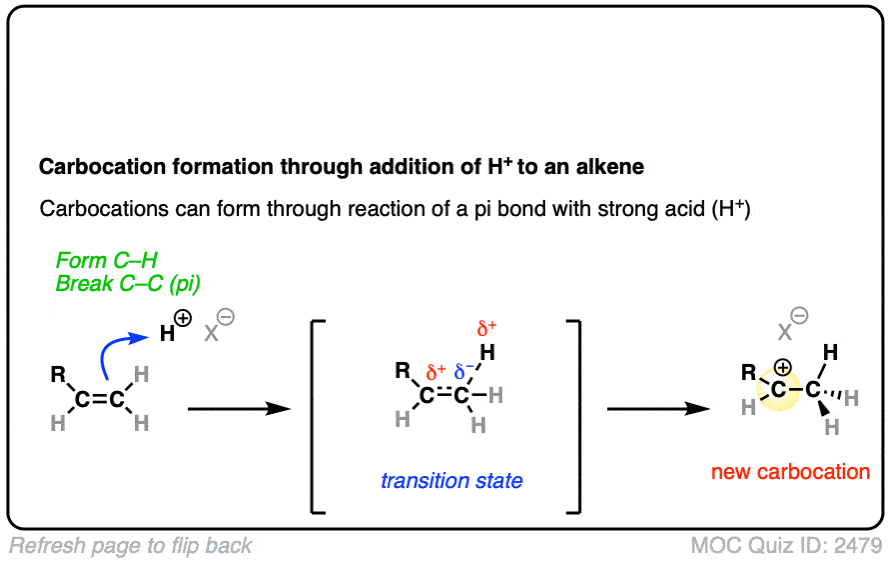
In both of these reactions, we start with a relatively low-energy starting material that passes through a high energy transition state en route to the carbocation intermediate.
In the second step, the carbocation then reacts with a Lewis base in solution to give a product where the carbon is again tetrahedral and sp3 hybridized.
A sketch of the reaction coordinates for each of these two reactions would look a little like this:
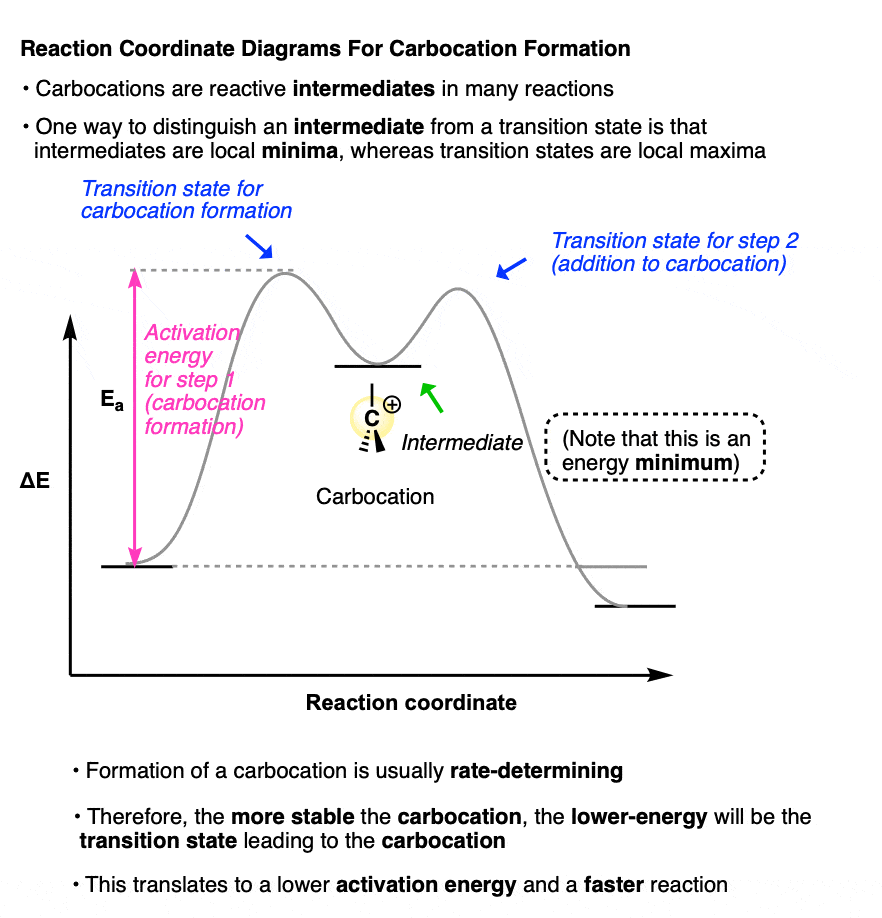
(Note that the carbocation intermediate occupies a local minimum on this reaction coordinate diagram; the two transition states are local maxima. )
Why bring this up?
Because the rate-determining step for each of these reactions is formation of the carbocation intermediate.
The more stable the carbocation intermediate, the lower in energy the transition state that leads to that carbocation, which translates into a lower activation energy and a faster reaction.
Therefore, if we understand the factors that govern the stability of carbocations, then it will also help us understand why certain reactions happen quickly whereas others do not!
3. Factors That Stabilize Carbocations – Substitution
The experimentally measured stability [Note 4] of carbocations shows the following trend:
Methyl (least stable) < primary < secondary < tertiary (most stable)
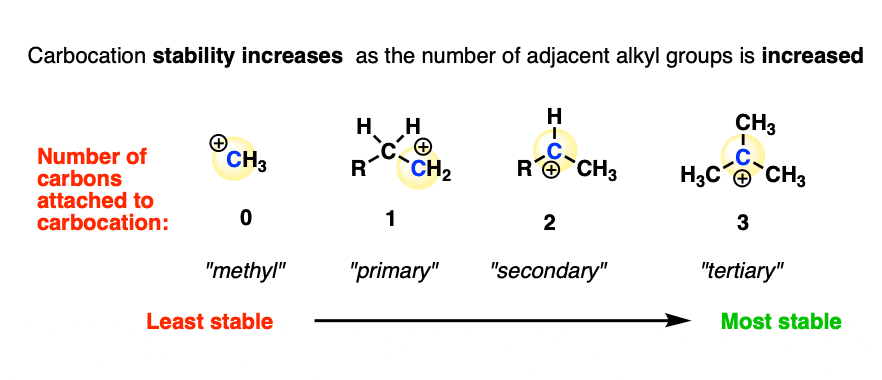
In other words, carbocation stability increases as C-H bonds are replaced with C-C bonds.
Why?
Being electron-poor, carbocations are stabilized through donation of electron density from neighboring electron-rich atoms.
As I’ve often told my students, “when you’re poor, it helps to have rich neighbors“.
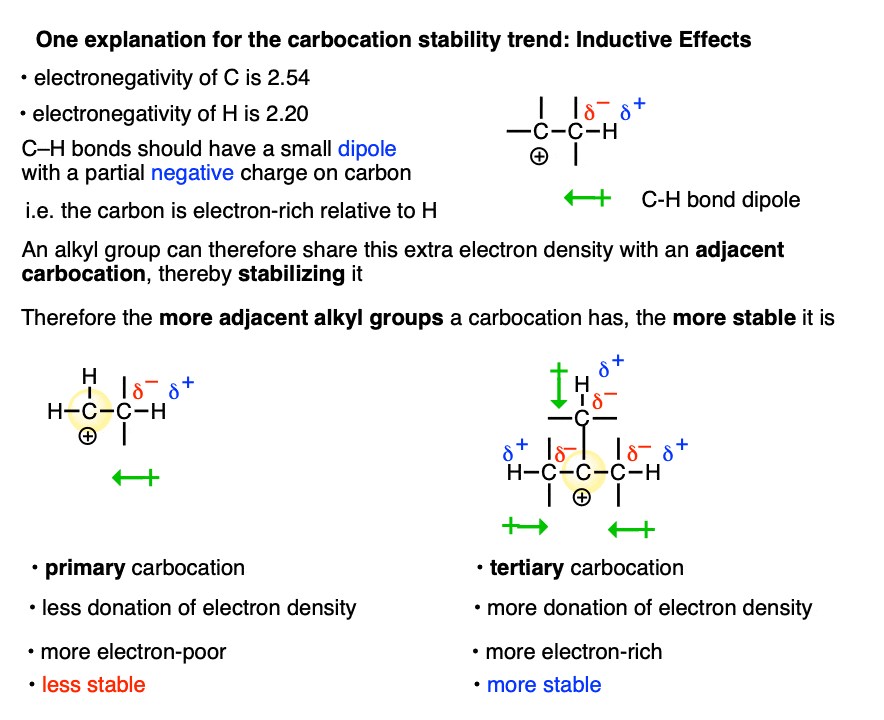
One way to rationalize this trend is through applying inductive effects.
Carbon is more electronegative (2.54) than hydrogen (2.20). So C-H bonds bear a small dipole whereby the carbon is partially negative and the hydrogen is partially positive.
The electron density from multiple C–H dipoles can add up. So the carbon of an alkyl group will have a small partial negative charge which can be donated to the adjacent carbocation, making it less electron-poor.
This isn’t possible when the carbocation is directly attached to H.
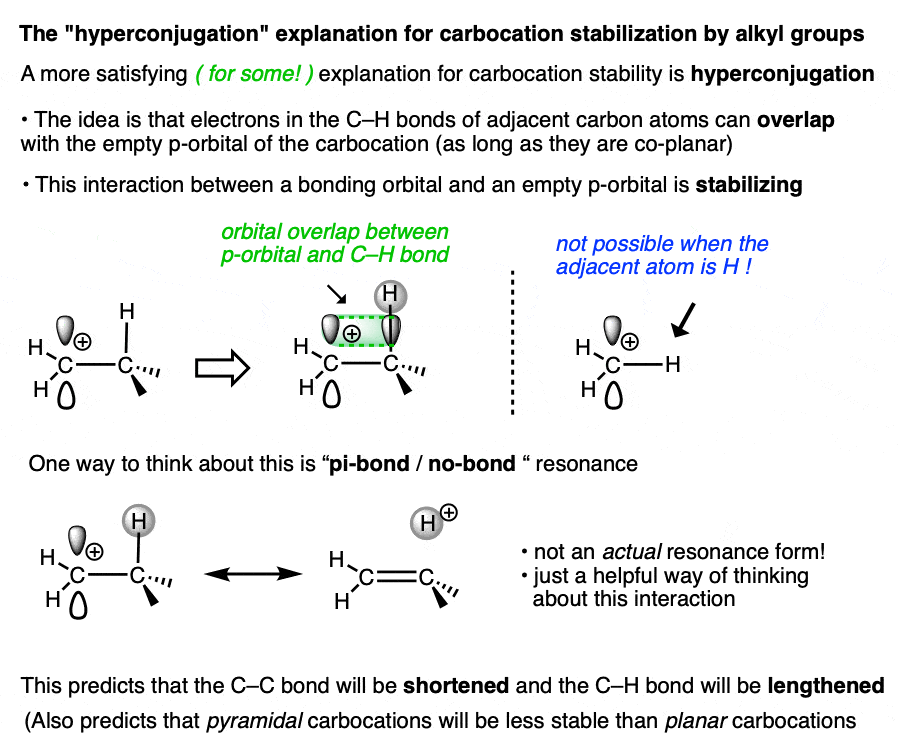
A more satisfying (to some! ) explanation comes from hyperconjugation.
Imagine lining up the two electrons in a C-H (or C-C) sigma bond with the empty p-orbital of the carbocation. Then imagine a form of “resonance” where those electrons are shared with the p-orbital to form a pi-bond (and H+).
Since electron density is being donated to an empty orbital, this should be stabilizing for the empty p-orbital (i.e. the carbocation).
We don’t have space in this article to go into all the implications of hyperconjugation in this article, but it does make some testable predictions that are borne out by experiment. [Note 3– the structure of the adamantyl carbocation is very instructive]
Whatever rationalization you prefer, the observed trend remains the same. The more substituted the carbocation, the greater its stability.
4. Factors That Stabilize Carbocations – Resonance
A good working principle in organic chemistry is that concentrated charge is generally more unstable (higher-energy) than dilute charge (lower-energy) [Note 5]
One place we’ve seen this before is in the concept of polarizability, where a big anion like iodide I(-) is more stable than a small ion like F(-) because the negative charge is spread out over a much greater volume. [This is the factor responsible for the greater acidity of H-I versus H-F and also the greater leaving group ability of I(-) ]
Another factor that results in this “spreading-out” of charges is “resonance”.
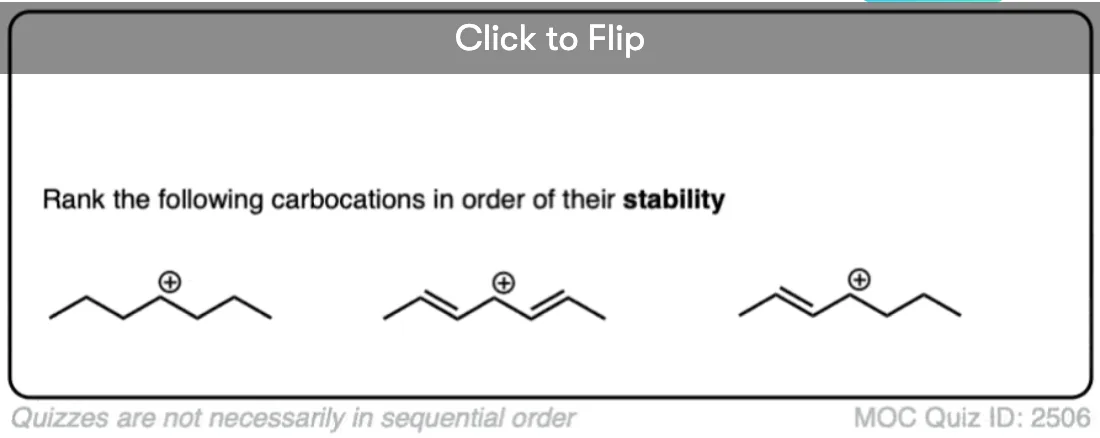
In a carbocation such as the n-propyl cation (CH3CH2CH2+) , the positive charge is localized to a single carbon.
But if a pi-bond is adjacent to the carbocation, we can then draw two resonance forms where the positive charge is either on C-1 or C-3.
In the resonance hybrid, the positive charge is shared between these two carbons, which will each have a charge density of +0.5 instead of +1. (This is what is meant by “resonance delocalization” – the charge has been smeared over multiple carbons)
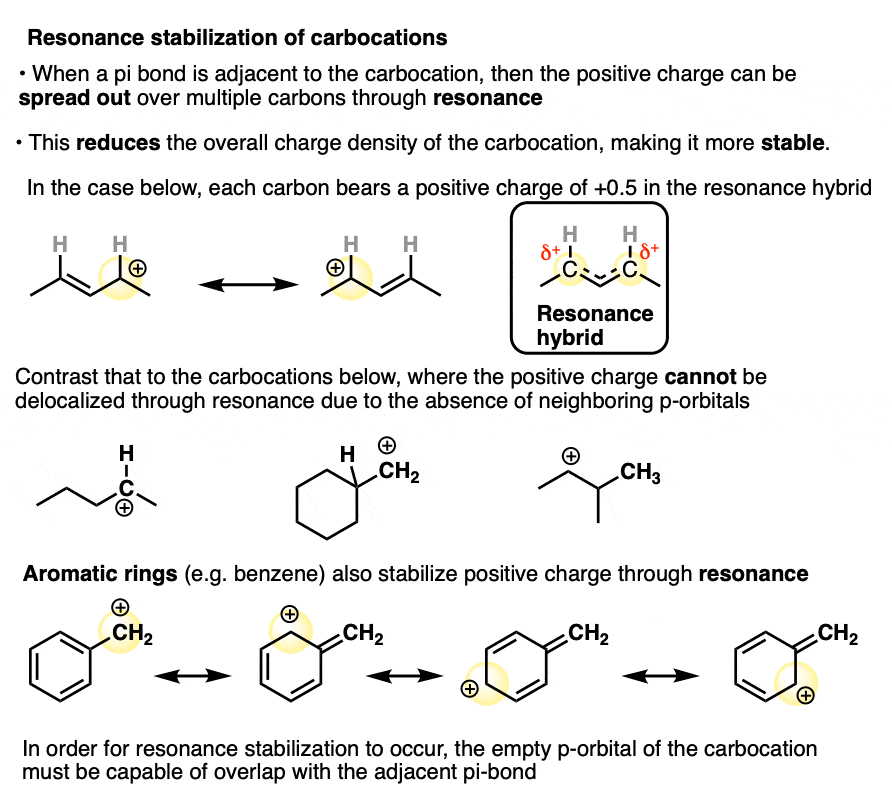
The greater stability of this carbocation (known as the allyl cation) relative to the parent propyl cation is borne out by numerous measurements. [Note 4]
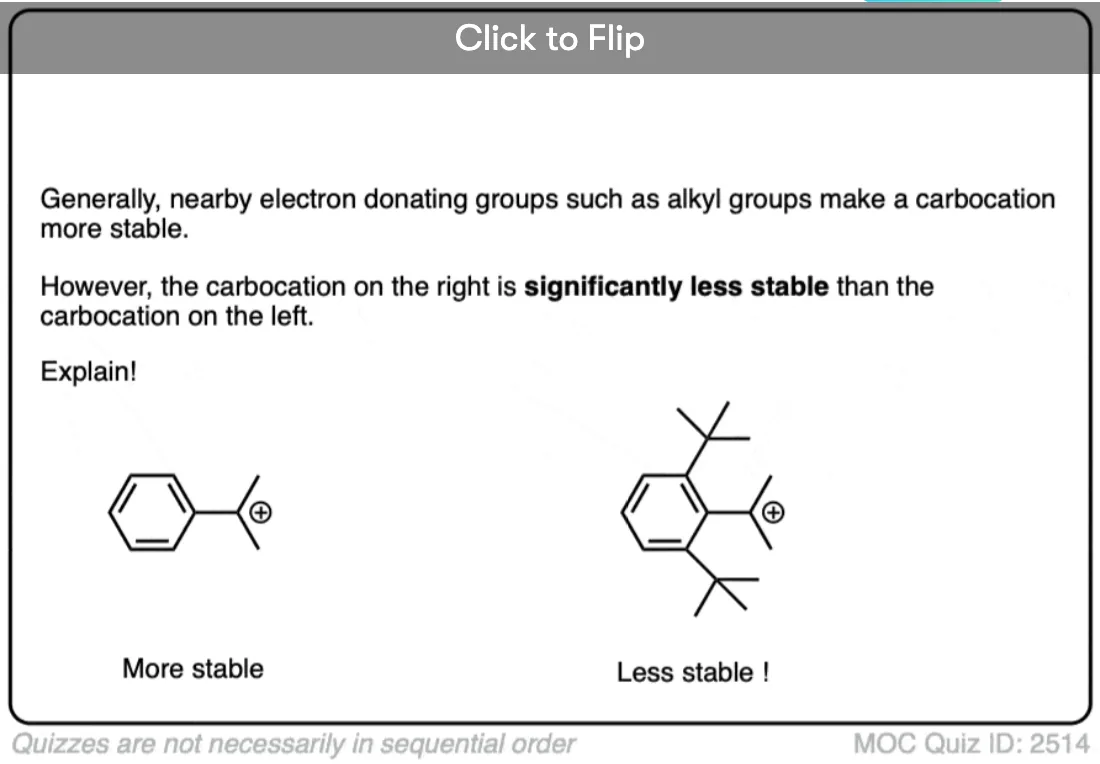
A similar effect is seen when a carbocation is adjacent to an aromatic ring such as benzene. The stability of this carbocation, known as the benzyl carbocation is considerably greater than that of a “normal” primary carbocation. (In fact the benzyl carbocation is roughly as stable as the t-butyl cation, Note 4)
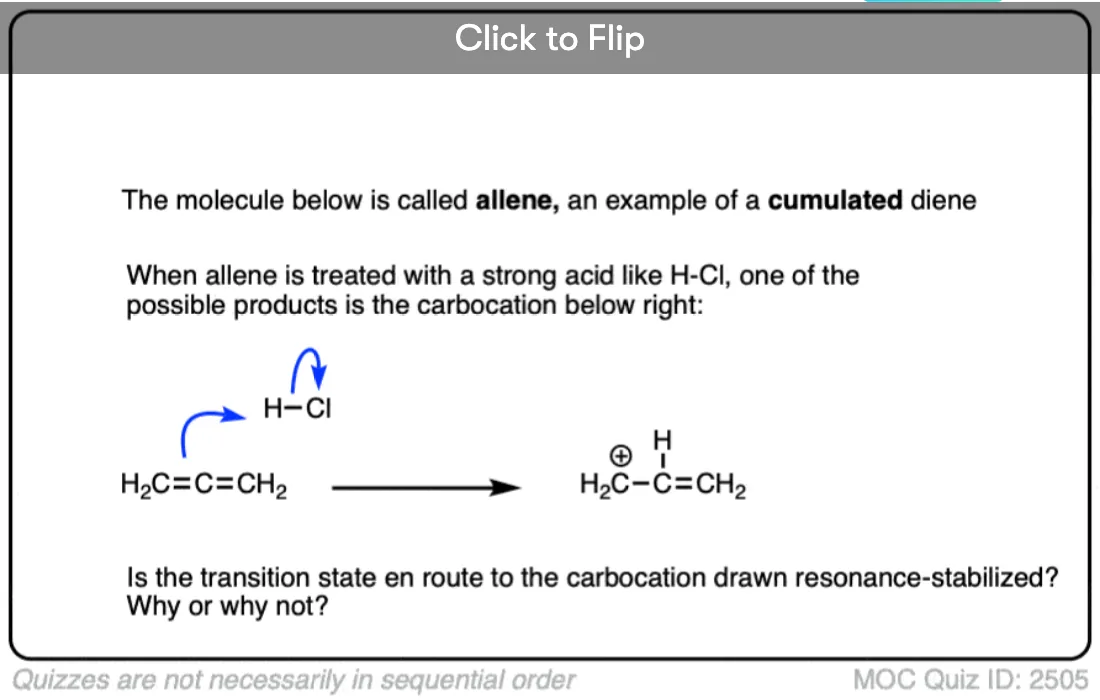
The stability of the carbocation tends to increase as the number of potential resonance forms increases.
For example, the “trityl” cation, Ph3C(+) is so stable that it forms a crystalline salt that can be put in a bottle and stored on a shelf indefinitely.
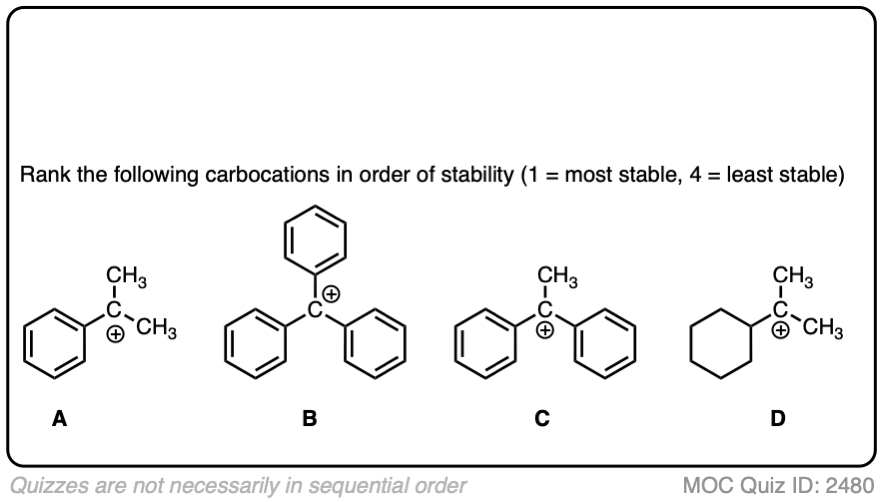 Click to Flip
Click to Flip
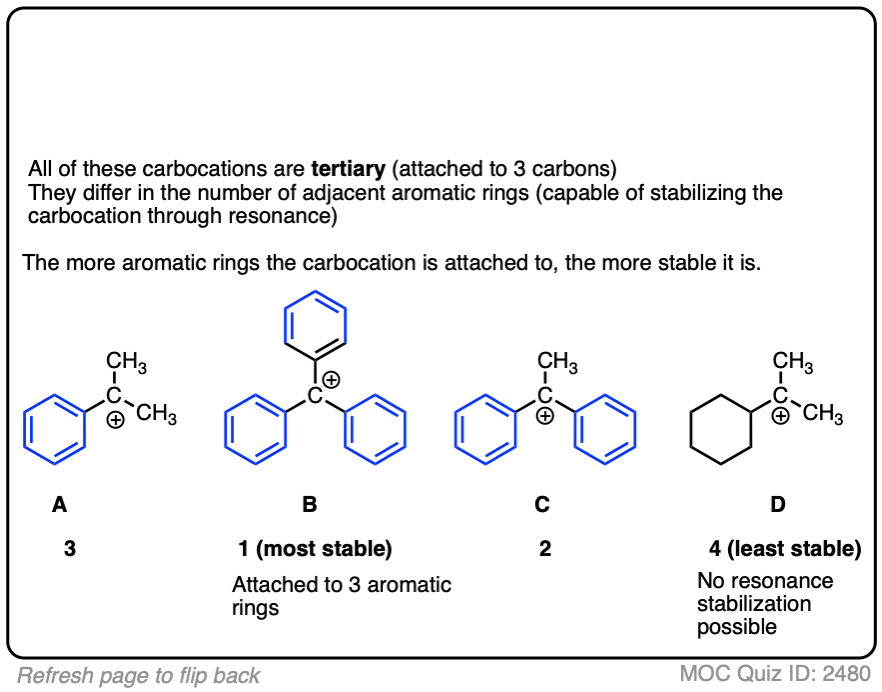
A word of warning. In order for a carbocation to be stabilized by resonance, the p-orbital of the carbocation must be able to overlap with the p-orbitals of the adjacent pi-bond. (See article – Conjugation and Resonance)
No overlap means no delocalization, which means no added stability.
Test your understanding of this concept by answering the quiz below.
 Click to Flip
Click to Flip
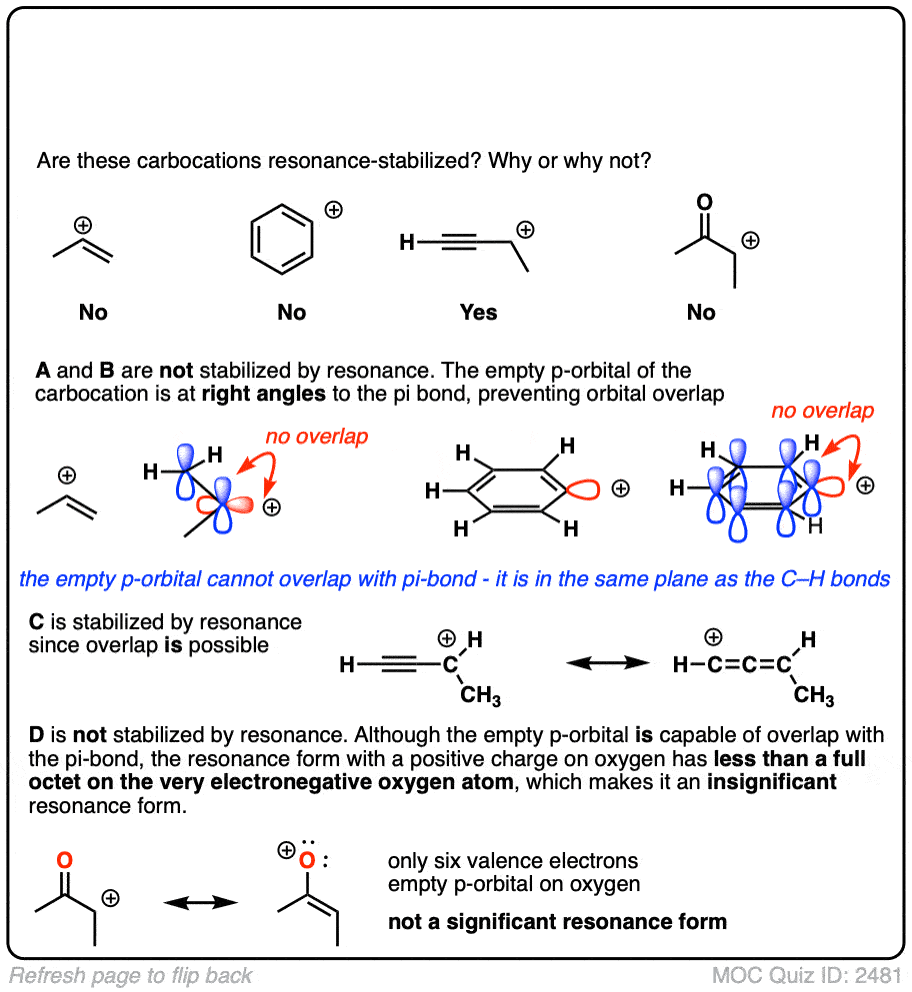
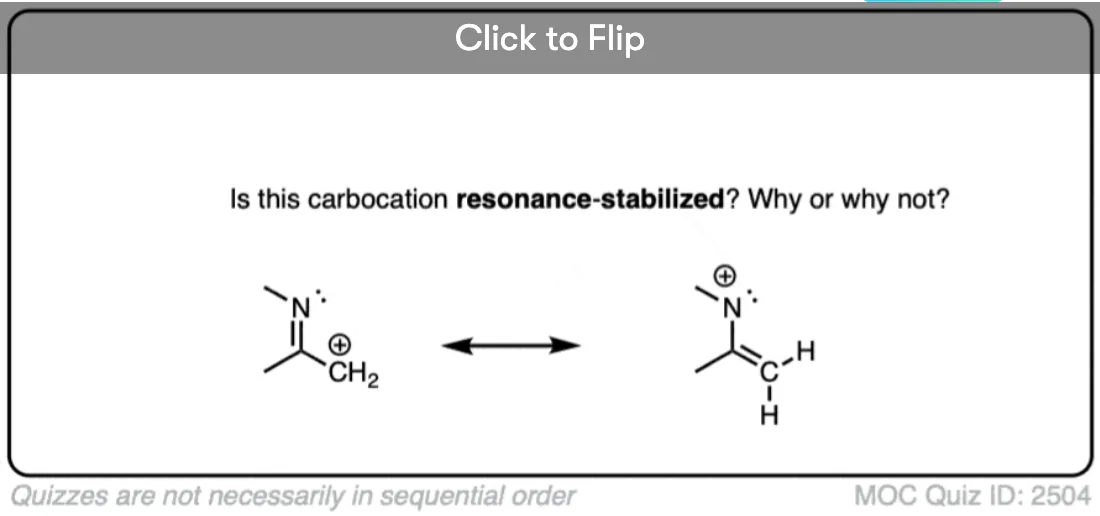
A common mistake is to think that carbocations directly attached to a pi-bond are resonance stabilized. This is not so! The pi-bond must be adjacent to the carbocation.
Another potentially counter-intuitive situation is when a carbocation is adjacent to a C=O (or C=N) bond. One might naively think that the carbocation is stabilized by resonance.
However, if you draw out the resonance form, you’ll find that in forming the C-C pi bond and breaking the C-O pi bond, you end up forming an electron-deficient species with only six valence electrons on oxygen. This “resonance form” is so high-energy as to make essentially no contribution to the resonance hybrid.
(Highly electronegative atoms like O are excellent at stabilizing negative charge, but they are terrible at stabilizing empty orbitals)
5. The Importance of Carbocation Stability In Reactions
Let’s return to the role of carbocations as intermediates in several key organic reactions.
The rate-determining step of the reaction below is protonation of the pi bond with H+ to give a carbocation intermediate.
Which of the two reactions do you think is faster?
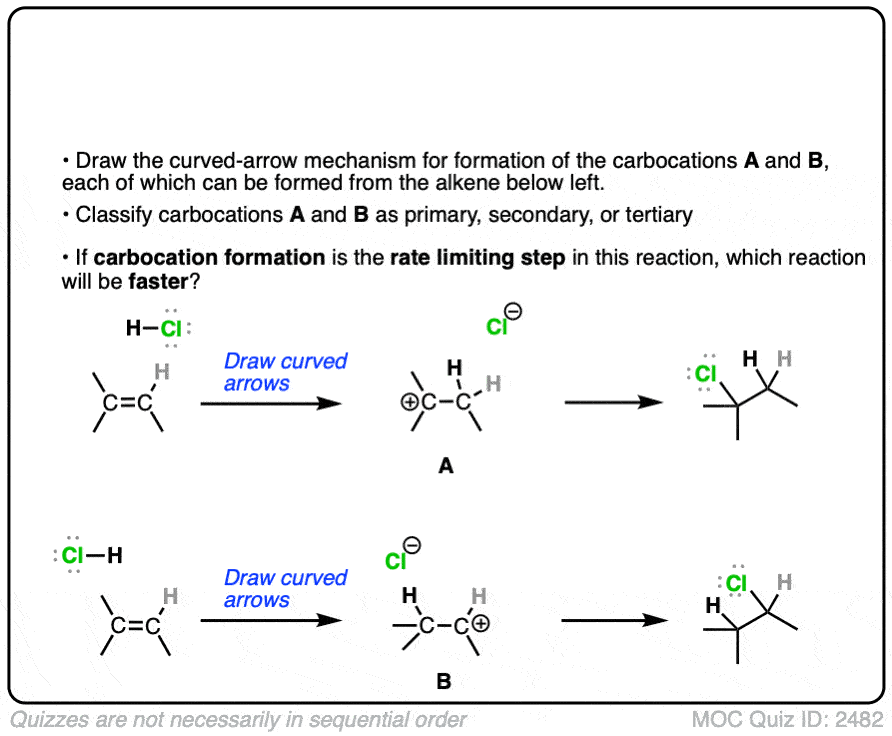 Click to Flip
Click to Flip
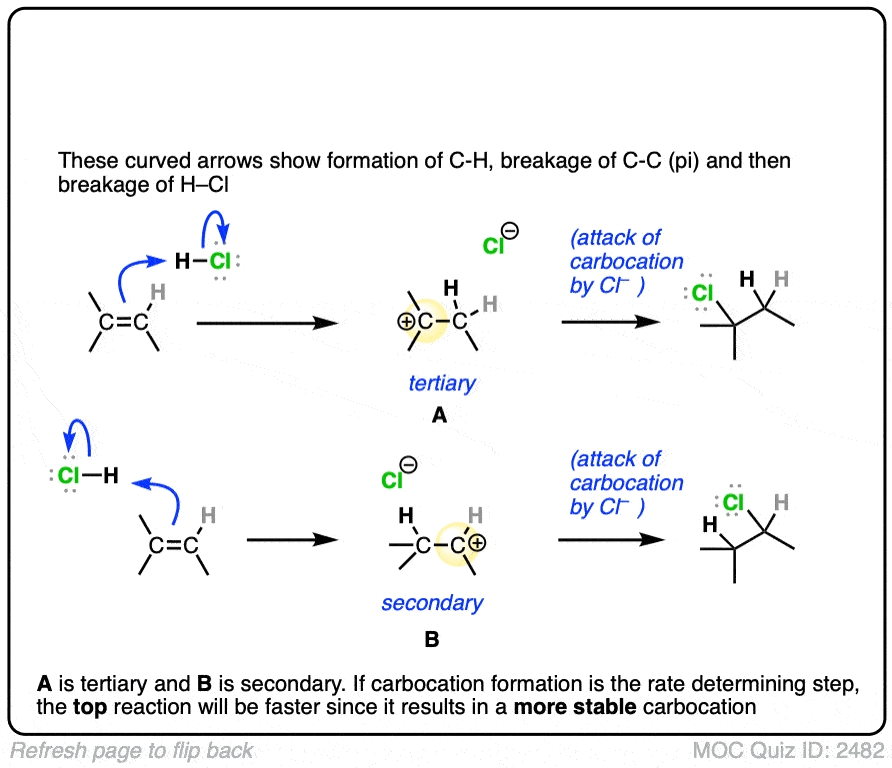
If you answered correctly, congratulations! You’ve shown you understand the reason behind Markovnikov’s rule! [See article – Markovnikov’s rule]
Let’s look at another one.
The rate-determining step of the reaction below is loss of a leaving group to form a carbocation.
Which of these two reactions proceeds faster?
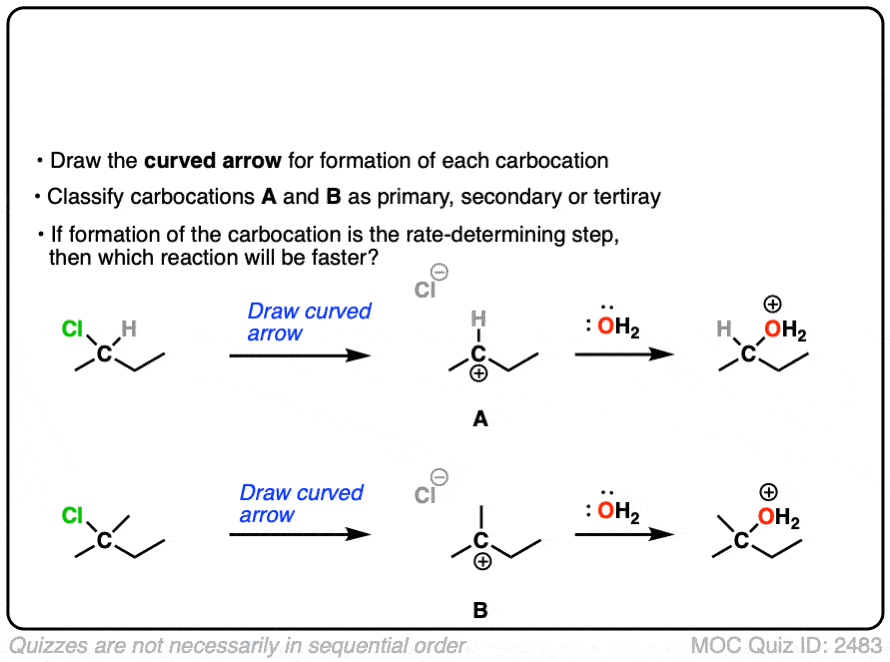 Click to Flip
Click to Flip
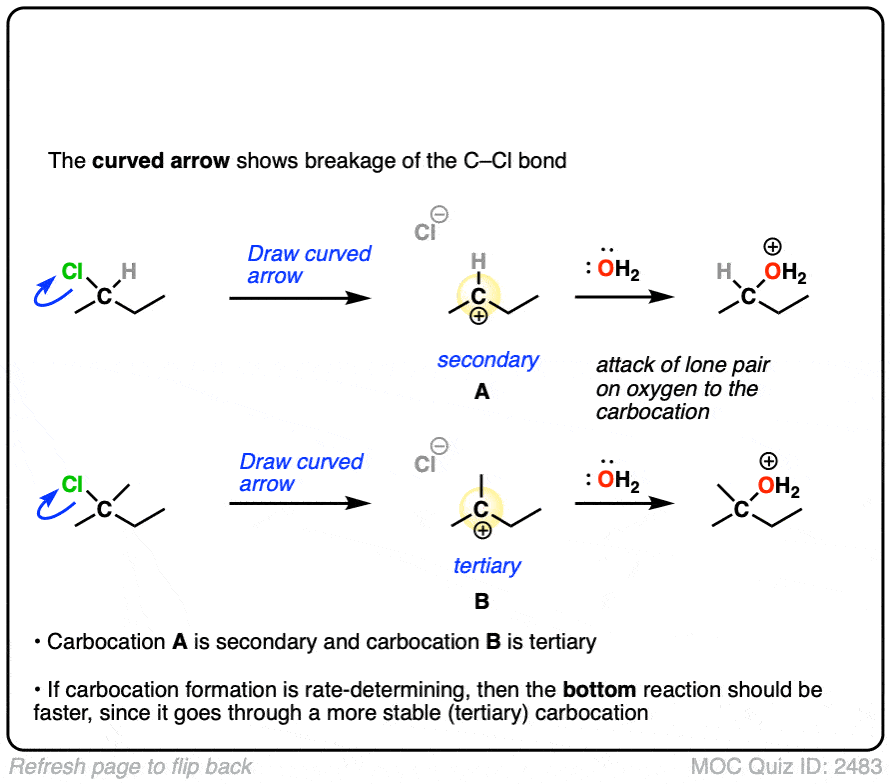
If you answered correctly, you’ve just shown an understanding one of the key factors that go into deciding whether a reaction will proceed via SN1, SN2, E1 or E2. (See article: SN1/SN2/E1/E2 – The Substrate)
6. Factors That Stabilize Carbocations – Adjacent Lone Pairs
There’s another factor that stabilizes carbocations that we haven’t touched on yet.
If a carbocation is formed adjacent to an atom bearing a lone pair (i.e. a Lewis base) then that atom can donate its pair of electrons to the carbocation, forming a new pi-bond in the process.
This is known as “pi-donation” (See article – Pi Donation)
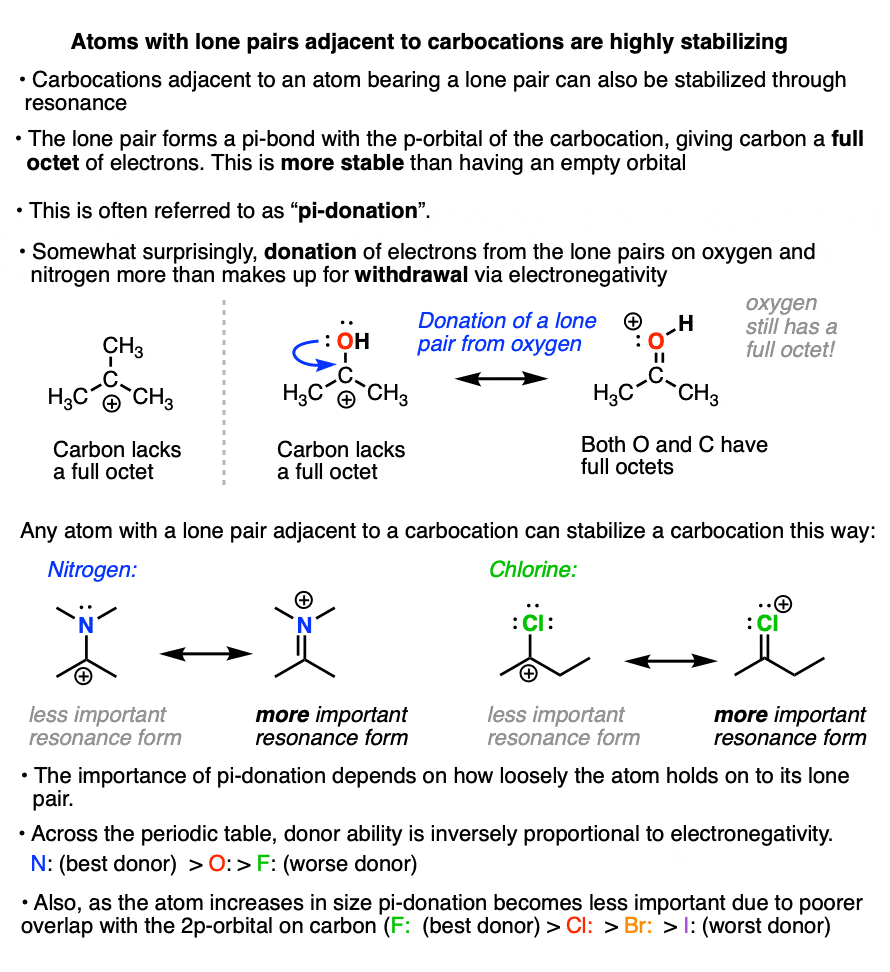
It might seem somewhat counter-intuitive that a highly electronegative atom like oxygen or nitrogen can stabilize a carbocation.
But the net result of pi-donation is that all atoms have a full octet, which is highly stabilizing. (Note that even though oxygen and nitrogen will bear a formal charge of +1, they still have a full octet of electrons! See article – How To Calculate Formal Charge)
The extent to which an atom’s lone pair can stabilize an adjacent carbocation is proportional to its basicity. The more basic the atom, the better the pi-donor.
Across a row of the periodic table, the ability to donate a pair of electrons is inversely proportional to electronegativity. So, all other factors being equal, a lone pair from nitrogen will be more stabilizing than a lone pair from oxygen, which is more stabilizing than a lone pair from fluorine.
Going down the periodic table, the ability of an atom to donate a pair of electrons is related to the ability of the atom’s valence orbital to overlap with the empty carbon 2p orbital.
Generally, as the orbital increases in size, orbital overlap (and pi-donation ability) will decrease.
Thus fluorine is a better pi-donor to carbon than chlorine, which is a better pi-donor than bromine, which is better than iodine (in iodine the valence orbitals are the 5p and 5s orbitals, which do not match up well with the smaller 2p orbital of carbon).
This is an article about carbocation stability and I want to keep it focused, but the contents of this particular section are extraordinarily important, and will come up again and again throughout organic chemistry. One application you’ll see later – Electrophilic Aromatic Substitution.
7. Which Factor is More Important – Substitution or Resonance?
So we have two factors that influence carbocation stability: substitution (primary, secondary, tertiary) and resonance.
So which is more important?
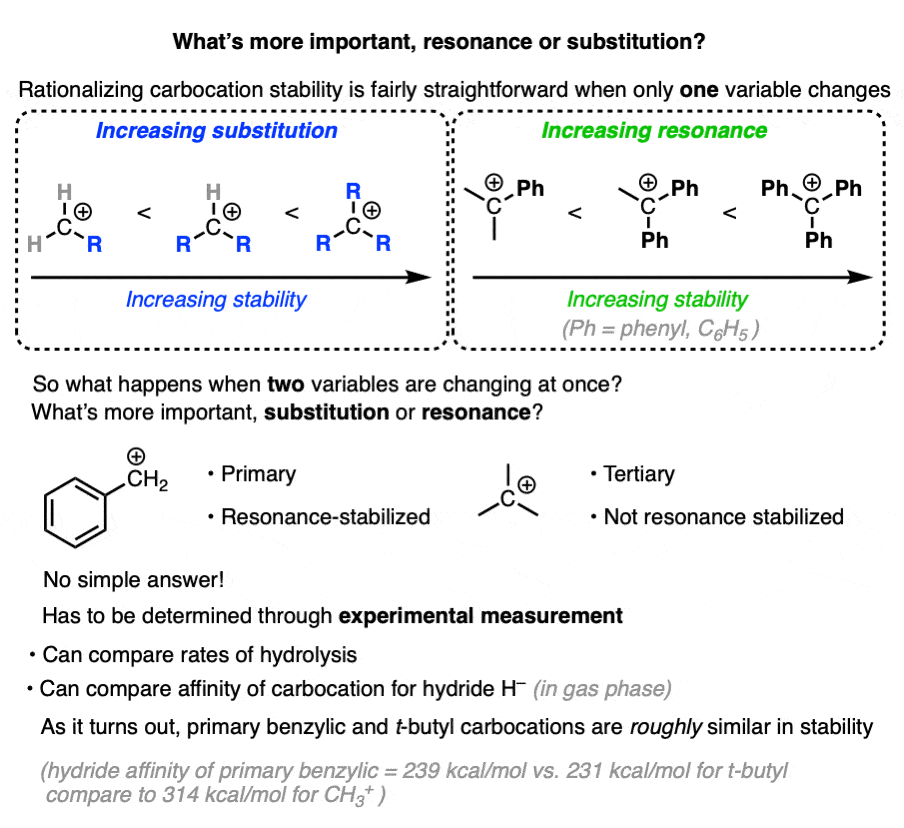
“they’re both important” is my non-answer to this question.
The actual way to answer this question is to look at experimental results.
One way to measure carbocation stability is to take a related series of alkyl halides and measure the rate of their hydrolysis in a polar protic solvent under conditions where SN1 would be likely.
(The advantage of this method is that it more closely approximates actual reaction conditions. One disadvantage is that it is really only applicable to carbocations that can form under SN1 conditions. )
Another method is to measure what is called hydride affinity – the energy released by the reaction of the carbocation with one equivalent of hydride ion (H–). (Although this can be applied to a greater range of carbocations, it is really only applicable in the gas phase).
The hydride affinity of a primary benzylic carbocation is about 239 kcal/mol versus 231 kcal/mol for a t-butyl carbocation, so these are quite comparable in energy.
A table with some values is below. See Note 4.
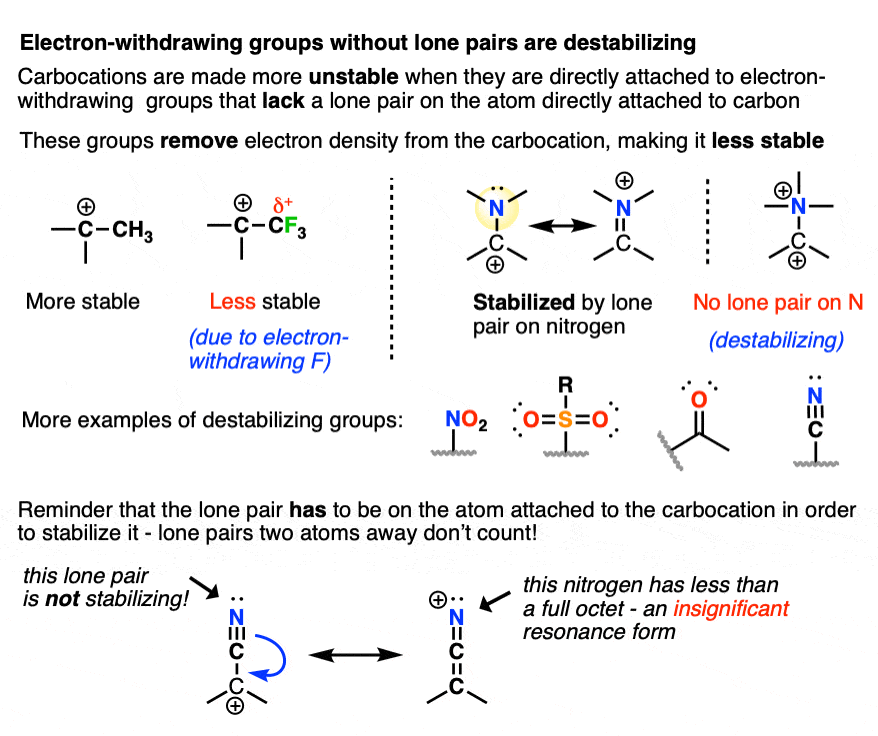
8. Some Factors That Destabilize Carbocations
Since electron-donating groups help to stabilize carbocations, it would make sense to expect that electron-withdrawing groups will destabilize carbocations.
As we touched on above, electronegative atoms with lone pairs like O, N, and F that are directly attached to a carbocation will actually help to stabilize carbocations through pi-donation.
I would not consider those “electron-withdrawing groups” for these purposes.
By “electron-withdrawing groups” I would include
- Carbon atoms attached to electron-withdrawing groups (e.g. CF3, CCl3)
- Electronegative atoms without lone pairs (e.g. NR3(+) )
- Atoms containing a pi-bond to a more electronegative atom (e.g. C=O, CN, NO2, SO2R, etc.)
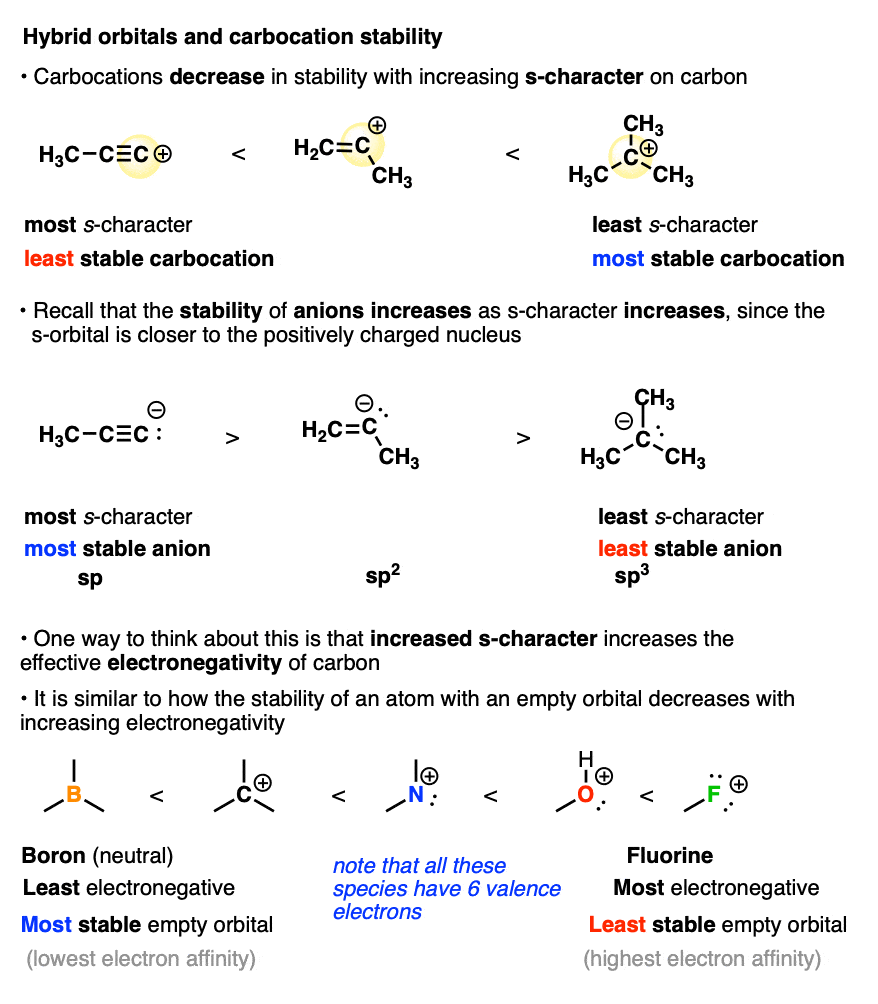
Another factor that destabilizes carbocations is the amount of s-character in the carbon atom.
You may recall that alkyne C-H bonds are particularly acidic because their sp-orbitals have 50% s-character, and since the s-orbital is closer to the positively charged nucleus, this helps to stabilize the negative charge of the conjugate base (a carbanion).
Well, when it comes to the stabilization of carbocations, this all gets thrown into reverse.
The more s-character the carbocation has, the closer that empty orbital is held to the positively charged nucleus. This has the effect of making the carbon nucleus have a greater effective electronegativity. Removing a pair of electrons to form a carbocation becomes increasingly difficult as the s-character increases. (This can be quantified by measuring hydride affinity or electron affinity)
Bridgehead carbocations are also particularly unstable due to the fact that they cannot attain the ideal trigonal planar geometry, [Note 6] as are carbocations on small rings (such as the cyclopropyl cation and cyclobutyl cation). [Note 7]
9. Some Special Cases
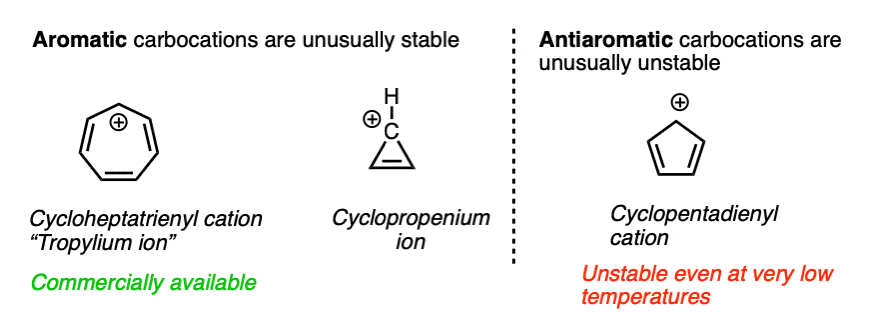
For the sake of completeness we should probably mention one last factor that is more of a second-semester topic but has a very large impact on the stability of certain carbocations.
Some molecules have a particularly stable property known as aromaticity. (See article – Rules For Aromaticity)
A related phenomenon called antiaromaticity is responsible for the unusual instability of certain compounds. (See article – Antiaromatic Compounds and Antiaromaticity)
The heptatrienyl (“tropylium”) cation, C7H7+ is aromatic and due to its unusual stability, forms a stable salt that can be put in a bottle and sold commercially. The cyclopropenium ion C3H3+ is also aromatic and unusually stable.
On the other hand, the cyclopentadienyl cation, which by all appearances should be resonance-stabilized, is actually anti-aromatic and has only a fleeting existence under carefully controlled conditions at very low temperatures.
10. Summary
Carbocations are positively charged, six-electron carbon atoms with an empty p-orbital. They are important intermediates in many reactions and are highly reactive towards Lewis bases.
Three key factors stabilize carbocations:
- First, they are stabilized by adjacent alkyl groups, which can donate electron density to the electron-deficient carbon atom.
- Secondly, they can be stabilized through conjugation with pi bonds, which allows the positive charge to be delocalized through resonance.
- Third, carbocations are stabilized by atoms with a lone pair capable of forming a pi bond.
Being electron-deficient, carbocations are destabilized by strongly electron-withdrawing substituents incapable of donating electron pairs (e.g. CF3, C=O). They are also destabilized if they are unable to attain the ideal trigonal planar geometry, which can happen in bridgehead carbocations. For similar reasons, carbocations that are part of highly strained rings such as cyclopropanes and cyclobutanes tend to be quite unstable.
Notes
Note 1. For most purposes, “carbocation” refers to a carbon with three bonds and an empty orbital, and in the vast majority of cases, this is the terminology that is used. There is a lot of inconsistency in the older literature as to whether these are “carbonium” or “carbenium” ions. The isolation of the CH5(+) species in extremely strong acid solution by the research group of George Olah at USC led IUPAC to define the term “carbenium ion” as a trivalent carbon and “carbonium ion” as a five-coordinate carbon. [Ref].
Note 2. Two prominent exceptions to this are the vinyl carbocation and the acylium ion.
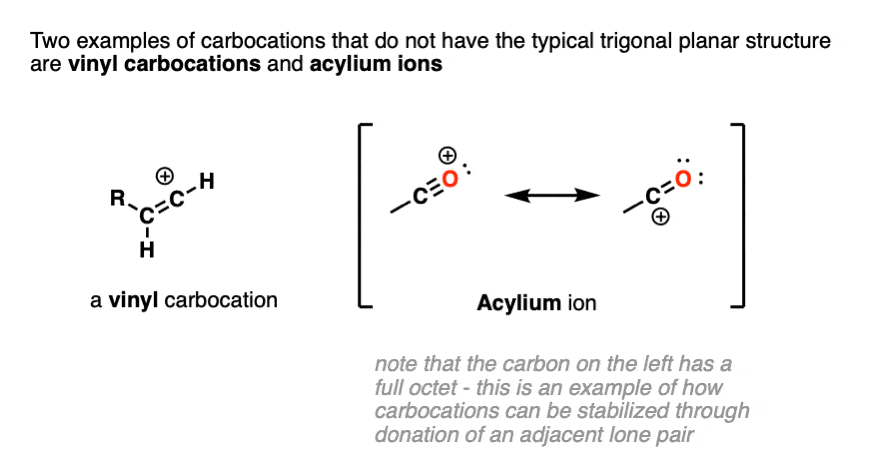
Note that the vinyl carbocation has a linear geometry and the central carbon is sp-hybridized.
The acylium ion is an excellent example of a carbocation that is stabilized through donation of an adjacent lone pair. (For examples of the acylium ion, see – The Friedel Crafts Alkylation and Acylation).
Note 3. The structures of several carbocations have been determined through X-ray crystallography.
For example, the structure of the t-butyl cation shows a trigonal planar structure (bond angles 120°) with C-C bond length of 1.44 Angstroms [Ref] which is about halfway between a typical C-C single bond (1.50 Å) and a C-C double bond (1.40 Å)
The structure of the adamantyl cation is particularly interesting. In adamantane, all C-C bonds are approximately the same length (1.53 Å). However, in the adamantyl carbocation, the carbon directly bonded to the carbocation becomes noticably shorter (1.43 Å) while the C-C bond that is able to overlap with the p-orbital of the carbocation is noticeably longer (1.608 Å).
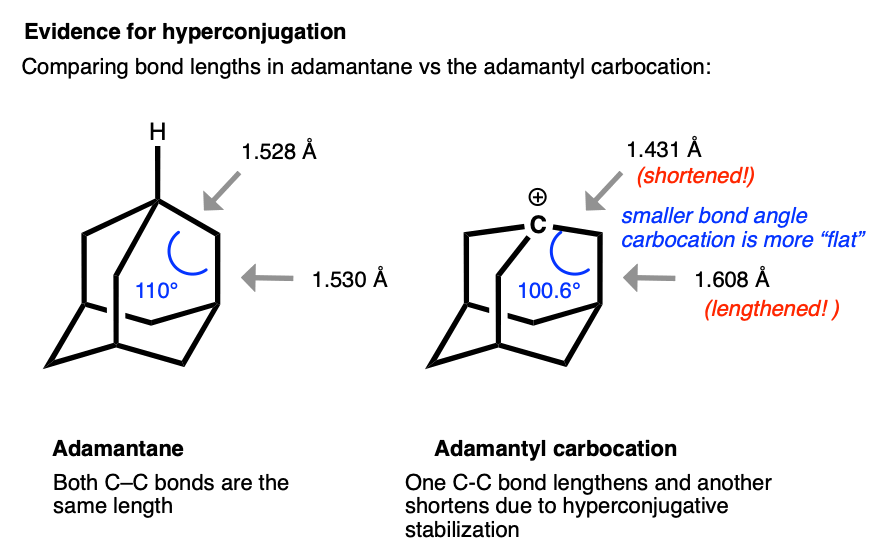
This is exactly what we would have predicted from using the hyperconjugation model for carbocation stabilization, where one C-C bond gains partial double bond character and another bond is weakened.
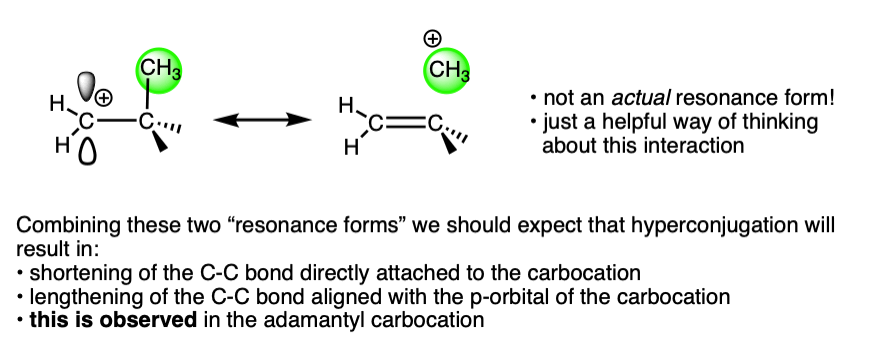
For a lot more detail on this, I highly recommend lecture 30 from Prof. David Evans’ Advanced Organic Chemistry 206 at Harvard, link here.
Note 4. One way to quantify the stability of carbocations is through measuring their hydride affinities, that is, the energy released upon reaction of the carbocation with a hydride ion. The hydride affinities of a large number of carbocations have been measured in the gas phase (where they have longer lifetimes).
A selection is provided below:
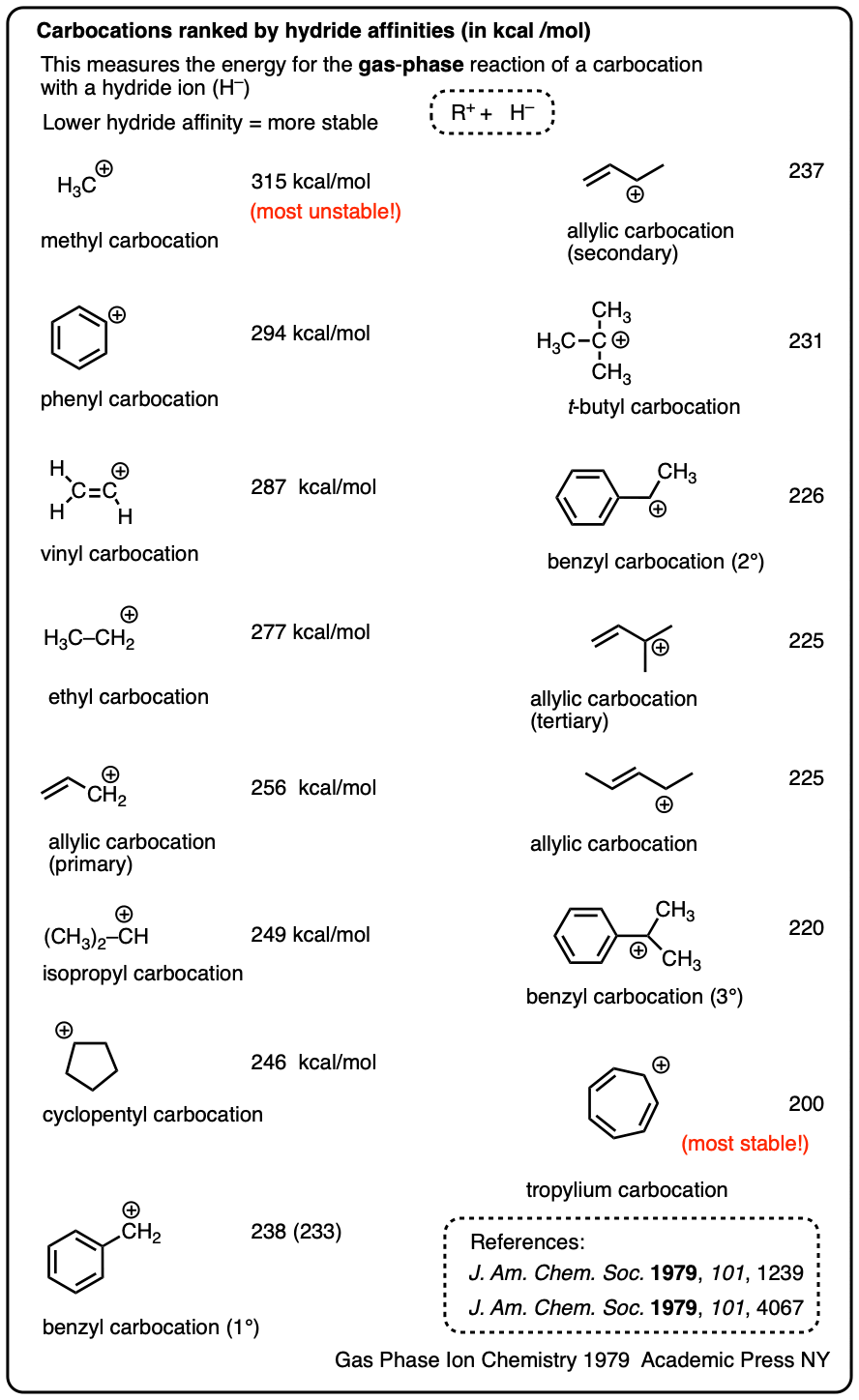
Solution-phase hydride affinities have also been measured for some of the more stable carbocations (gas phase hydride affinities in parentheses)
- Ph3C(+) : 96 kcal/mol
- Ph2CH(+): 105 kcal/mol
- PhCH2(+): 118 kcal/mol (238 kcal/mol)
- Tropylium ion: 83 kcal/mol (200 kcal/mol)
Note 5. Carbocation formation is easier in solvents with a high dielectric constant such as water or carboxylic acids (e.g. acetic acid, formic acid).
Note 6 One class of tertiary carbocations that are particularly unstable are carbocations that are constrained in a bridgehead. Ring strain prevents these carbocations from adopting a trigonal planar geometry.
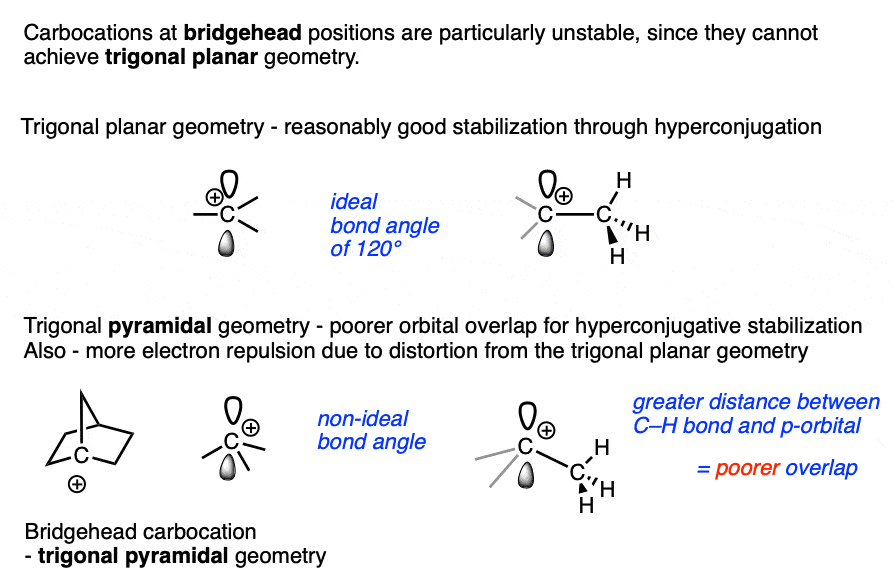
Note 7. Carbocations that are part of small rings such as cyclopropyl and cyclobutyl carbocations are also unusually unstable.
Note 8. An alternative method of measuring carbocation stability is through measuring rates of hydrolysis in a nucleophilic solvent. However, as this graphic from Evans shows, these two measures do not always correlate well…

Quiz Yourself!
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.

Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading
References
- 100 Years of Carbocations and Their Significance in Chemistry1
George A. OlahThe Journal of Organic Chemistry 2001 66 (18), 5943-5957
DOI: 10.1021/jo010438x
A historical perspective from Prof. George Olah, who won the Nobel Prize in Chemistry in 1994 for “contributions to carbocation chemistry”. - Stabilities of carbocations in solution. 14. An extended thermochemical scale of carbocation stabilities in a common superacid
Edward M. Arnett and Thomas C. Hofelich
Journal of the American Chemical Society 1983, 105 (9), 2889-2895
DOI: 10.1021/ja00347a060
This paper provides a table of the stability of 39 carbocations, measured calorimetrically by heat of formation (ionization in superacid from the corresponding carbinol). - X-ray Crystal Structures of Carbocations Stabilized by Bridging or Hyperconjugation
Thomas Laube
Accounts of Chemical Research 1995 28 (10), 399-405
DOI: 10.1021/ar00058a001
Very interesting review from Prof. Laube comparing and contrasting the structures of various carbocations, including very clear evidence for hyperconjugation in the adamantyl carbocation. C-C bond shortened to 1.431 from 1.528 . C-C lengthenend to 1.608 from 1.530. 100 deg angle vs 110 deg angle - Carbonium Ions. I. An Acidity Function (C0) Derived from Arylcarbonium Ion Equilibria
C. Deno, J. J. Jaruzelski, and Alan Schriesheim
Journal of the American Chemical Society 1955, 77 (11), 3044-3051
DOI: 10.1021/ja01616a036
Carbocation stability can also be expressed in pKR+, which is defined in the paper. The carbocations studied in this paper are all relatively stable arylcarbonium ions. - Hydride affinities of carbenium ions in acetonitrile and dimethyl sulfoxide solution
Jinpei Cheng, Kishan L. Handoo, and Vernon D. Parker
Journal of the American Chemical Society 1993, 115 (7), 2655-2660
DOI: 10.1021/ja00060a014
Carbocation stabilities can also be expressed in terms of hydride affinity (R+ + H– -> RH). The carbocations studied in this paper are also fairly stable arylcarbonium ions, as these are measured electrochemically in either DMSO or acetonitrile. - Photoelectron spectroscopy of methyl, ethyl, isopropyl, and tert-butyl radicals. Implications for the thermochemistry and structures of the radicals and their corresponding carbonium ions
A. Houle and J. L. Beauchamp
Journal of the American Chemical Society 1979, 101 (15), 4067-4074
DOI: 10.1021/ja00509a010
Table III in this paper has heats of formation for the basic alkyl cations (methyl, ethyl, isopropyl, and t-butyl), but in the gas phase. The numbers agree with intuition from solution-phase experiments; stability increases from methyl -> ethyl -> isopropyl -> t-butyl. - Quantitative preparation and enthalpy of rearrangement of the sec-butyl cation
W. Bittner, E. M. Arnett, and M. Saunders
Journal of the American Chemical Society 1976, 98 (12), 3734-3735
DOI: 10.1021/ja00428a072
This paper describes a novel calorimetric method for measuring the heat of isomerization of the sec-butyl cation to the tert-butyl cation in superacid solution. Using this, the value obtained is 14.5 ± 0.5 kcal/mol. - Stability of Alkyl Carbocations
Thomas Hansen, Pascal Vermeeren, F. Matthias Bickelhaupt, and Trevor A. Hamlin
Chem. Commun. 2022, 58, 12050-12053
DOI: 10.1039/D2CC04034D
A theoretical study on carbocation stability trends. From the paper: “we quantitatively established a generally overlooked driving force behind the stability of carbocations, namely, that the parent substrates are substantially destabilized by the introduction of substituents, often playing a dominant role in solution. This stems from the repulsion between the substituents and the C–X bond.“ - The Story of the Wagner-Meerwein Rearrangement
Ludmila BirladeanuJournal of Chemical Education 2000 77 (7), 858
DOI: 10.1021/ed077p858
An article of historical interest on the development of understanding of the Wagner-Meerwein rearrangement of carbocations.
Hello James, thanks for the clear explanation.
A question about the carbocation. We say that the more stable carbocation reacts with the nucleophile faster. Are we connecting the thermodynamic stability to the kinetics here?
Hi Chenglin, I am assuming you are referring to the image under section 5.
The *overall* kinetics of these reactions will be dictated by the formation of the carbocation, which is the rate-limiting step. Generally the more stable the carbocation, the lower will be the the activation energy for its formation (“late” transition state) and the faster the overall rate of reaction.
It should not be interpreted to mean that there is a relationship between the stability of the carbocation and the rate of the *second* (i.e. fast addition of nucleophile to carbocation) step.
Cyclopropenyl cation being aromatic can be stored in bottle.but is it possible to store cyclopropyl methyl cation as it is more stable.
Which carbocation is more stable
Cyclopropyl methyl cation or cyclopropenyl cation
Cyclopropyl methyl. Cyclopropenyl quickly rearranges to allyl cation.
thank you sir your articles are very hepful and they helped me to understand my archj-enemy O-chem better
THANK YOU!!!!
From gas phase dissociation energies, the tert butyl carbocation is about 7 kcal/mol more stable (232 kcal/mol) than the benzyl carbocation (238 kcal/mol) but substituent effects can greatly change these numbers. Source: March’s Advanced Organic Chemistry 5th ed. Table 5.2 page 224.
Hi Sir!
I Hav a question.
Which one is more stable
Benzyllic cation or CH2(+)-cyclopropane??
That is an EXCELLENT question and the data contradicts somewhat. Cyclopropylmethyl cations are generally considered to be more stable than benzyl. I’m looking for a better reference than just March 5th ed. p. 222, but the references therein are to good, but somewhat obscure, reviews. Hydrolysis rates suggest cyclopropylmethyl cations are more stable.
However another way to answer that is to look at 13-C NMR to determine the chemical shift of the carbocations. The more negative the chemical shift, the more unstable it is. The 2-cyclopropyl carbocation has a chemical shift of -86.8 ppm and the 2-phenylpropyl cation has a chemical shift of -61.1 indicating that the phenyl group is better at stabilizing. See https://pubs.acs.org/doi/10.1021/ja00731a026
Will a primary alkyl halide be able to undergo an SN1 reaction if it is stabilised by a neighbouring oxygen atom? Or will the carbocation still be too unstable to react in this way?
Depends on what you mean by “neighboring”. If it’s directly attached to the same carbon (such as H3C-O-CH2Cl) then the oxygen will easily form a pi bond with C and the Cl- will be displaced easily. For an example see benzyloxymethyl chloride. (these things tend to be unstable)
If by neighboring you mean an C=O on the carbon adjacent to the carbon bearing the carbocation, then this will be unstable.
No matter where I surf the net… I’ll always end up here.Your way of teaching is just amazing…hats-off
I hope this continues!
Is it possible for a non-adjacent atom with a lone pair to stabilize a carbocation? There’s a question in Brown’s Organic Chemistry 8th edition that asks why CH2CHCOOH (https://image.ibb.co/cdThRJ/structure_1.png) + HCl forms CH2ClCH2COOH rather than CH3CHClCOOH which would be expected due to the ordinarily greater stability of the secondary carbocation. I’m wondering if you can get the carbon backbone curling around upon itself (https://image.ibb.co/d6eBLd/structure_is_this_what_s_happening.png) so the lone pair on the hydroxyl oxygen can help to stabilize the primary carbocation?
Sorry, an extra carbon snuck into those molecular models. The correct structure of the starting material is (https://image.ibb.co/nOFkfd/structure_1.png) and the correct structure of my proposed stabilized carbocation is (https://image.ibb.co/cxTSty/structure_is_this_what_s_happening.png).
I suppose this could also be a contributing resonance structure (https://image.ibb.co/hj8Vfd/structure_contributing_resonance_structure.png).
Well what could happen is intramolecular nucleophilic attack to give a ring of some kind. “Anchimeric assistance”.
Thank you so much, sir! You’re metaphor on money and electrons made the concept so much easier! More powers to you!
Are there answers to the “apply the concept” questions?
Dear James:
I have no problem with -NH2, -OH since we establish in EAS that they are electron donating in general. But i don’t understand why halides should stabilize the carbocation. It is said in all textbooks that the mistmatch between 3p and 2p atomic orbitals results in a diminished resonance delocalization, and winning of the inductive effect for halides, making them electron-withdrawing. So it removes electron density and creates a bigger positive charge in the intermediate of aromatic reaction. but we have the same halide and the same carbocation here (Except its not in a ring) but its now donating whats the difference? Thanks!
So if you have a secondary carbocation that has a little bit of resonance stabilization and a tertiary carbocation (with no resonance stability), which is more stable? (My textbook says the secondary with resonance…)
Hello, can you please tell me which is more stable carbocation…(CH3)3C or (C2H5)3C ?
If the no of carbon atoms increases in an alkyl group…Its +I effect will decrease or increase ?
Perhaps a sight increase in stability for tri-ethyl. The rate of ionization of the corresponding alkyl halides would tell you.
I’m wondering if there an answers to the Apply questions at the bottom to check ourselves???
Where are the ans of the examples?
(CH3)2–C+ —COOH , can this resonate ? …i mean theres pi sigma and +ve charge conjugate system ….but yet i was doing a question and this wasn’t the answer.
So why cant it resonate please answer
The resonance form would end up with less than a full octet on oxygen, which is extremely unstable. It’s not a significant resonance form.
No resonance stabilization. If you break the C=O bond to form a C=C bond you will leave behind a oxygen with only 6 electrons –> highly unstable.
I don’t find any article on destabilization of carbanion
Some factors that stabilize negative charge:
https://www.masterorganicchemistry.com/2012/02/27/7-factors-that-stabilize-negative-charge-in-organic-chemistry/
Hi!
I was just wondering, which would be more stable then between a tertiatry carbocation and a carbocation stabilized by resonance? (like in #2?)
Thank you! Great article!
Hey James….
Why an intermediate with positively charged oxygen is less stable than a carbocation
There are two types of intermediates with positively charged oxygen. The intermediate where oxygen has a full octet is OK (and generally speaking more stable than a carbocation). Intermediates where oxygen have less than a full octet are very unstable, because a very electronegative atom (oxygen) with less than a full octet will have tremendous potential energy (and thus instability) for pulling electrons toward the nucleus.
By positively charged oxygen I mean an electron with 6 electrons. It’s a more electronegative element so there will be MUCH greater electron-affinity pulling electrons toward the nucleus. Very unstable situation.
Hi James,
I would like to pick your brain a little bit. I have been banging my head against the wall with this one and I don’t seem to figure it out.
Let’s say you have two secondary amines. In the first one, one substituent is a propargyl (prop-2-yn) group and the second one is methyl. In the second amine, one substituent is an (indol-2-yl)methyl group (amine is bonded through a methylene group to the position 2 of indole) and the second one is methyl. Now, in both cases a secondary carbocation is formed next to the amine nitrogen. Which one should be more stable?
In both cases the nitrogen stabilizes the positive charge via lone electron donation, but what about the alkynyl and indolyl moiety? I suppose they should both have stabylizing effects via delocalization but which one would stabilize the charged species more? What about the inductive effects? Should they play some significant roles in this case? I am so confused…
Hi James,
Thanks for a neat explanation. I am not an organic chemist and I have a question about stabilization of carbocations. Do you think it is probable to stabilize a carbocation by putting it next to sth that can stabilize it? Like solvation effects, or some negatively charged species?Like electrostatic stabilization?
In all these 3 examples, carbocation is stabilized via intramolecular effects, how about intermolecular stabilization?
Thanks.
Carbocations are relatively stable in superacid solution. That’s how George Olah studied them, and it helped him to win the Nobel Prize. http://www.nobelprize.org/nobel_prizes/chemistry/laureates/1994/olah-lecture.html
Thanks much.
Organic chemistry has always been the most exiting and beautiful subject for a geek like me and all these articles inspire me to go more in depth of this subject.
amen to that brother
Hi there, why is a secondary allylic carbocation more stable than a tertiary carbocation? Thank you.
Hi Monica
There’s several factors that are not always easy to judge by just looking at them – we need to do experiments. For example we know that carbocations increase in stability going from primary to secondary to tertiary.
We also know that carbocations increase in stability if they are resonance stabilized.
However – what factor is more important? substitution pattern or resonance?
There is no way to figure this out just by looking at it. We have to do experiments (and we do!) [by measuring “ionization rates”]
Those experiments tell us that secondary allylic carbocations are slightly easier to form than ordinary (non resonance stabilized) tertiary carbocations.
Why is that? Good question (and this is where it can get complicated). Probably the fact that there is more electron density being donated from an adjacent p orbital than there is from the [hyperconjugation] C-H bonds adjacent to the tertiary carbocation.
These factors can be in delicate balance. If I can make an analogy, it’s a bit like sports teams. What’s more important in football, to have a good offence or a good defence? Well, they’re both important. How do we know the relative importance of each? By playing a LOT of games and trying to figure it out by looking at the data. Thankfully with chemistry one mole of material gives us the chance to play 6x 10^23 “games” so we can figure this out pretty reliably.
hope this helps! James
Hi,
I was wondering if you could post the answers to these sample problems please? I’m doing them, but I have no way of checking if it’s right.
Thank-you,
TA Nguyen
Hi, in each case the first carbocation is more stable. This applies the 3 factors we learned that stabilize carbocations.
tertiary carbocation more stable than secondary
allylic (resonance stabilized) carbocation more stable than non resonance stabilized carbocation
carbocation adjacent to atom with lone pairs (oxygen) more stable than carbocation not adjacent to atom with lone pairs.
Hope this helps
James
hello…i have a question can we say that the resonanse factor is more effective factor except of when the aromaticity is endangered?
Can you please tell me the stability order of tertiary,benzyl and allyl free radical?
Try looking at the strength of tertiary, benzyl, and allyl C-H bonds. Since C-H bond strengths measure homolytic cleavage, then you will then get the stability of the radicals. The weaker the C-H bond the more stable the radical.
Hi! Thanks for your help : )
I have a question. Would a secondary carbocation be considered more stable than a primary carbocation bonded with a halogen? It’s on a practice test and I’m a little confused o_O
Hard to say without seeing the exact example, but my guess is that the latter situation would be more stable, since the halogen can donate a lone pair and every atom on the molecule can have a full octet. This is a more stable situation than a free carbocation where there is an empty orbital.
First of all ,thanks for explaining this so well.
But, can u please tell me , generally which effect counts more, Inductive or Hyperconjugation?
Like for example, if you have ethyl carbocation and if you have 2 methyl propane carbocation (primary carbocation) which will be more stable?
It’s far more powerful to look at stabilization from the perspective of hyperconjugation, but it’s far easier to explain it from the perspective of inductive effects. If you are a major in a chemistry program I would take the time to learn hyperconjugation and apply that to your studies. It’s a very powerful concept.
can u please explain why 1,3,5 hept-triene carbo cation (+ on sp3 carbon) is more stable than triphenyl carbo cation??
Aromaticity. See: https://www.masterorganicchemistry.com/2017/02/23/rules-for-aromaticity/
This was so much help! Organic chem is a pain, are there more explanations of other orgo subjects?
like spectroscopy, I really need help on that and could use a good website like this one
Thanks.
Had a hard time finding bare-bone explanation of cation stability. Thanks! Definitely bookmarking your website.
Thanks. I’m now studying for my Organic Chem exam next week and this is really helpful for my studies
I have only a little problem . well, which counts more, the resonance stabilisation or if its primary or secondary Carbon? Due this fact, which is more stable, +CH2-CH=CH2 or CH3CH(+)CH3? Thank you in advance xD
Good question. In the examples you cited, the resonance counts more.
Thank you very much, this has saved my life. Appreciate it.
actually my main ques was about pinnacol pinnacolone rearrangement.
H+ attacks on that OH which yields a more stable carbocation
so which O should it attack?
OH OH
I I
CH3—C—C—-C2H5
I I
CH3 C2H5
oops the server omitted the spaces in the compound which messed it all up..
it is
(CH3)2–C(OH)————-C(OH)(C2H5)2
out of ch3ch2ch2+ and ch3ch2+ which is more stable carbocation
both are primary but
the former one has a bulkier alkyl group and hence more inductive effect
and the latter one has more no of alpha hydrogen and hence more no of hyperconjugative structures..
both the reasons are clashing……!!!!!
we expect the first one out of intuition but how can we forget the fact that hyperconjugaion is more dominant tha inductive effect?
Both are primary carbocations; they will have very similar stabilities. The propyl carbocation can rearrange through a hydride shift to give a secondary carbocation.
so the conclusion is that propyl carbocation is more stable….
bcz it can rearrange
No – once it’s rearranged, we’re discussing a different carbocation entirely.
What about the “bent or umbrella bond”? Don’t you think that bent bond participate in the stability of carbocations?
Can you add carbocation shift as well to make this complete.
Discussed in the series on rearrangments.
Oh man it is totally awesome. You told every thought tat we have while studying! And my every doubt is gone now !!n!n!n!
Bro, you make this shit easy. You are seriously awesome!
this article rly helped me alot !! m glad u posted it :D
Good article!
Great article.
Good explanation one can eaisly understand by reading this article
Glad you found it helpful.
can you please explain that if I have a benzyl carbocation and a t-butyl carbocation
which will be more stable
1st has stability due to benzyl resonance
and 2nd has 9 possible hyperconjugative structures
please answer
Dear Jeetesh! You should know that resonance is more pronounce than hyperconjugation and will stabilize the cation more as compare to hyperconjugation.
yes Iqbal,resonance is dominating mainly…..bt here it has been found that t-butyl is more stable…..
Kushal, do you have a reference for that?
Morrison boyd says this . (http://www.chemicalforums.com/index.php?topic=33527.0)
As one of the commenters says, small changes in substitution can tip the balance either way.
I do not have an experimental reference, but in “Electron Flow In Organic Chemistry” Paul Scudder claims on page 65 in his “carbocation stability ranking” chart that “tertiary cation” is more stable than “benzyl cation” (the benzyl shown being primary, i.e. two H’s).
I’d imagine that a tertiary benzyl cation, the sp2 carbon bearing a phenyl group and two alkyl groups, should certainly be more stable than a tertiary cation bearing three alkyl groups (both have two hyperconjugations, however the aromatic pi donor beats the third hyperconjugation).
A secondary benzyl cation vs tertiary alkyl cation would be a little more ambiguous.
Dear all, carbocation stability could be inferred from the hydride affinity in gas phase. Please see Advanced Organic Chemistry: Part A 5th edition by Carey and Sundberg, Table 3.10., page 303. The lower the hydride affinity, the more stable the carbocation. (239 kcal/mol for benzylic cation VS 237 kcal/mol for t-Butyl cation)
I understand Rich’s explanation. It’s satisfying. Thank You.
This is because the phenyl groups are not in a single plane and for resonance to occur, the R groups should lie in the same plane. This distortion of shape to the compound happens due to steric hinderance of the phenyl groups in the compound
Its an exceptional case, t-butyl carbocation is more stable …..
Benzyl carbocatian is more stable because of delocalization of charge…
very simple way to understand chemistry
information about organic chemistry
To explain science in simplest way is an art.you have done it!
thanks!
good tutorial.