Alkyne Reactions
Oxidation of Alkynes With O3 and KMnO4
Last updated: June 2nd, 2026 |
Oxidation of Alkynes with Ozone and KMnO4
- As we’ve seen previously, alkenes (olefins) can be oxidized to carbonyl compounds (aldehydes, ketones, carboxylic acids) with ozone and KMnO4. (See article – Alkene Reactions – Ozonolysis)
- Alkynes can also undergo oxidative cleavage with ozone and KMnO4.
- The products depend both on the structure of the alkyne and the reaction conditions.
- With internal alkynes R-C≡C-R , ozonolysis (or KMnO4) gives two carboxylic acids, R-CO2H.
- With terminal alkynes, R-C≡C-H, ozonolysis produces a single equivalent of carbon dioxide (CO2) and a chain-shortened carboxylic acid.
- Additionally, reaction conditions can be modified such that C-C bond cleavage does not occur and 1,2-diketones are formed.
Two oxidation reactions of alkenes that do not translate well to alkynes are the reaction of alkynes with OsO4 (dihydroxylation) and epoxidation with peroxyacids (e.g. m-CPBA). While alkynes do react with these reagents, the reaction products are complex, and we will not dwell on them except perhaps to gawk at the results in the footnotes at the bottom of this article.
[summary image]
Table of Contents
1. Oxidative Cleavage of Alkynes via Ozonolysis
- We’ve seen that alkenes (olefins) can be oxidized to carbonyl compounds (aldehydes, ketones, carboxylic acids) with ozone and KMnO4. (See article – Alkene Reactions – Ozonolysis)
The next logical question is: what about alkynes? Can we cleave them too? Not that you’d really want to in most circumstances, saying as alkynes are a wonderful blank canvas for organic synthesis and can be turned into a huge array of useful functional groups…. but if you had to, could you?
It turns out that the answer is yes!
The products that are obtained depends on the structure of the alkyne and the reaction conditions.
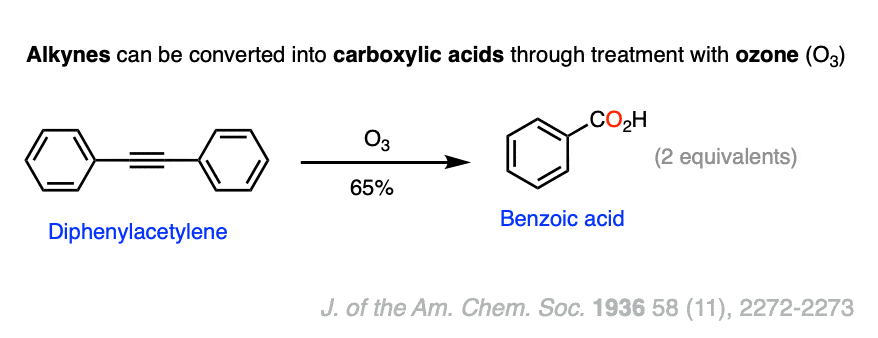
Internal alkynes, R-C≡C-R , can be cleaved via ozonolysis to give carboxylic acids. When the alkyne is symmetrical, this gives two identical carboxylic acids.
A prime example is the ozonolysis of diphenylacetylene, which gives benzoic acid.
Terminal alkynes, R-C≡C-H, also undergo oxidative cleavage. In this case, the terminal carbon bubbles out of the reaction mixture as carbon dioxide (CO2) and we are left with a chain-shortened carboxylic acid.
For example, 1-hexyne becomes pentanoic acid plus carbon dioxide:
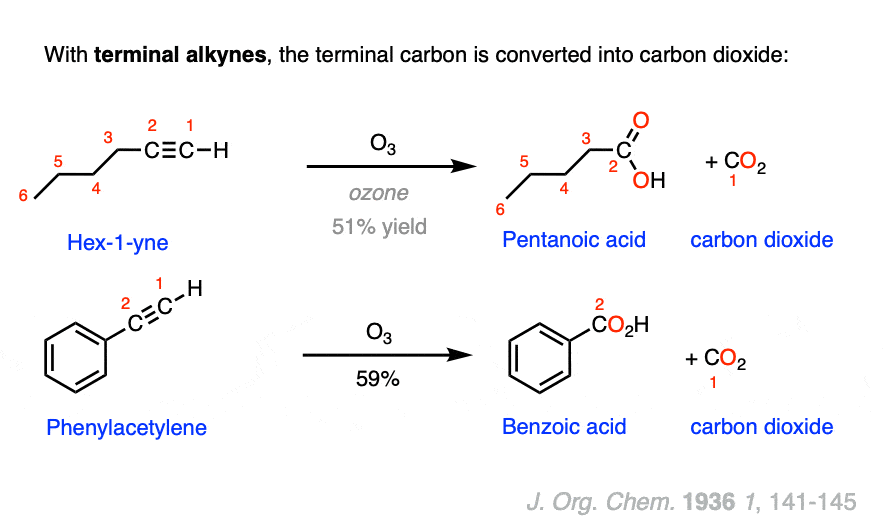
The original product of ozonolysis would be formic acid, HCO2H, but in the presence of ozone the C-H bond is oxidized further to give carbonic acid, CO(OH)2 . Whether during ozonolysis or in a open can of Diet Coke, carbonic acid is in equilibrium with CO2 and H2O, and gradually CO2 is lost to the atmosphere.
2. Cleavage of Alkynes With Permanganate (KMnO4)
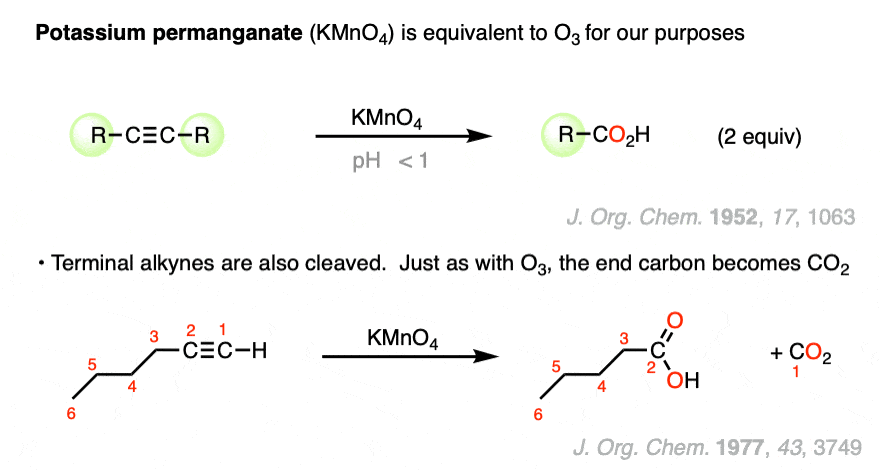
Under certain conditions potassium permanganate can also cleave carbon-carbon triple bonds.
For example, it is reported that KMnO4 under either strongly basic or strongly acidic conditions will break apart internal alkynes to give carboxylic acids.
As with ozonolysis, terminal alkynes are converted to the chain-shortened carboxylic acid and one equivalent of CO2.
3. Formation of 1,2-Diketones From Alkynes
As noted previously, forming carboxylic acids from alkynes, while occasionally useful, seems like a waste of two good C-C pi bonds that could otherwise be converted into something more interesting.
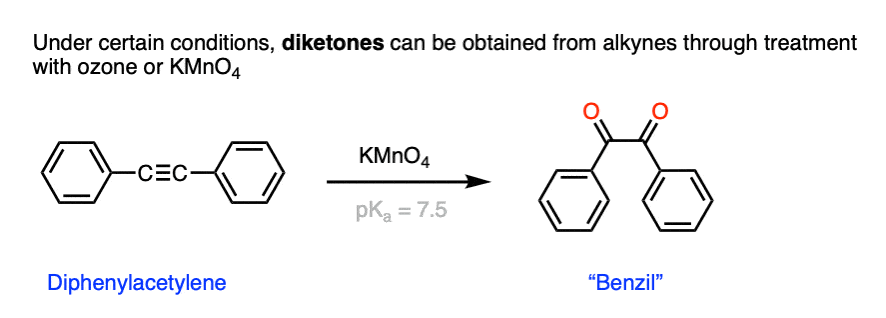
Thus, I am happy to report to you that under certain condtions, C-C triple bonds can be directly converted into 1,2-diketones.
Here, for example, is the reaction of phenylacetylene with KMnO4 to give benzil.
A number of other oxidants (e.g. RuO4) can also carry out this conversion, as does OsO4. [Note 1]
Notes
Some highlights from chapter 13 of Patai’s “Chemistry of Triple Bonded Functional Groups” Part 1, supplement C.
Note 1. On OsO4. Here’s what can happen if alkynes are treated with OsO4. They perform double addition (note that this is a proposed structure, not a conclusive proof) which can then be hydrolyzed to give a diketone.
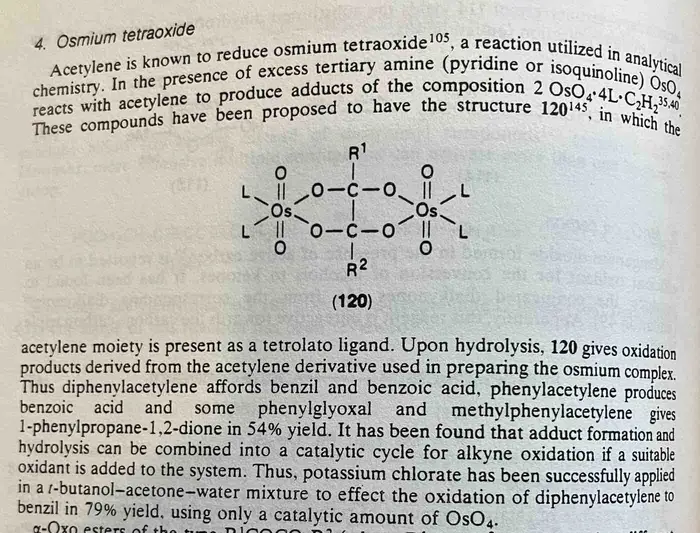
Note 2. The reaction with m-CPBA is even more interesting. Epoxidation of an alkyne gives rise to a transient, very unstable (due to antiaromaticity – see Antiaromatic Compounds And Antiaromaticity) oxirene, which quickly rearranges to an alpha-keto carbene. The carbene can then perform C-H insertion reactions on adjacent alkyl groups, such as t-butyl.
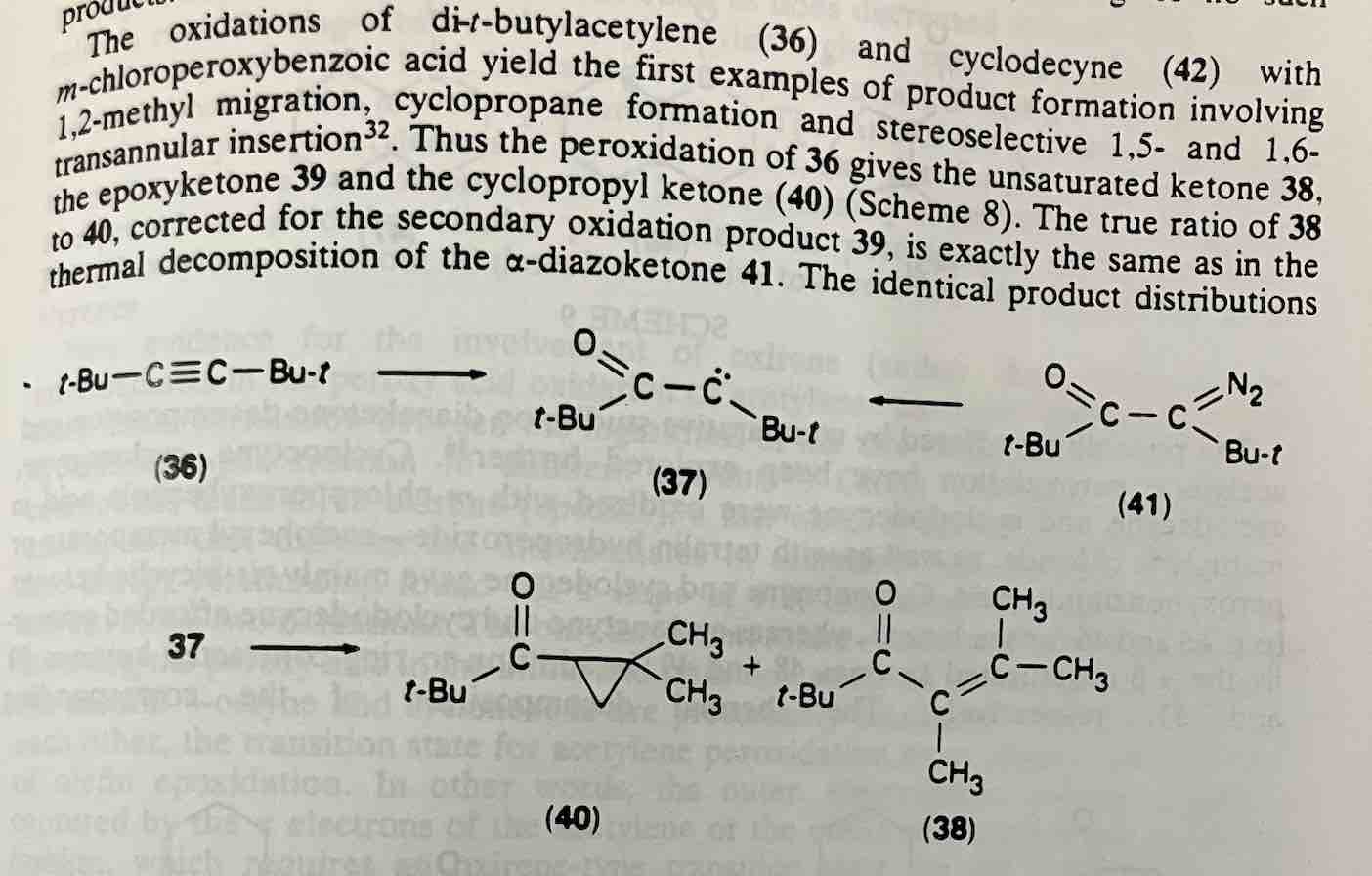
Reactions with cyclic alkynes are even more wild, with transannular C-H insertions to form various bicycles. See JACS 1973, 95, 3284.
Quiz Yourself!
[quizzes]
(Advanced) References and Further Reading
Ozonolysis of alkynes is not commonly performed and tracking down good examples required more than the usual amount of detective work.
Good leading reference is in Patai’s Chemistry of Triple Bonded Functional Groups” Supplement C Part 1 chapt 13, p. 523-524. (Laszlo I. Simandi).
- THE OZONIZATION OF TRIPLE BONDS
CHARLES D. HURD and ROBERT E. CHRIST
The Journal of Organic Chemistry 1936 01 (2), 141-145
DOI: 10.1021/jo01231a002
One of the first published studies of the ozonolysis of triple bonds, with yields in the 45-60% range. - Transformations of alkynes to carboxylic acids and their derivatives via CC bond cleavage
Shivalinga Kollea and Sanjay Batra
Org. Biomol. Chem., 2016,14, 11048-11060
DOI: 10.1039/C6OB01912A
A historical review on carbon-carbon triple bond cleavage and reactions, including ozonolysis, KMnO4, and many other reagents. - The Isolation of 1,2-Diketones from the Ozonization of Disubstituted Acetylenes
Thomas L. Jacobs
Journal of the American Chemical Society 1936 58 (11), 2272-2273
DOI: 10.1021/ja01302a053 - Ozonolysis of Unsymmetrical Acetylenes
PHILIP S. BAILEY, YUN-GER CHANG, and W. W. L. KWIE
The Journal of Organic Chemistry 1962 27 (4), 1198-1201
DOI: 10.1021/jo01051a017 - Ozonolysis of Alkynes—A Flexible Route to Alpha-Diketones: Synthesis of AI-2
Joshua L. Alterman, Dua X. Vang, Marissa Roghair Stroud, Larry J. Halverson, and George A. Kraus
Organic Letters 2020 22 (19), 7424-7426
DOI: 10.1021/acs.orglett.0c02182
A relatively recent application of alkyne ozonolysis, to obtain 1,2-diketones. - Reductive trapping in the ozonolysis of diphenylacetylene
S. Jackson and L. A. Hull
The Journal of Organic Chemistry 1976 41 (20), 3340-3342
DOI: 10.1021/jo00882a037
The authors study the ozonolysis of diphenylacetylene and propose a mechanism for the formation of carboxylic acids similar to that for the ozonolysis of alkenes. - Ozonization and Oxidation of Stearolic Acid to 9, 10-Diketostearic Acid
N. A. Khan and Melvin S. Newman
The Journal of Organic Chemistry 1952 17 (7), 1063-1065
DOI: 10.1021/jo50007a024
This study by Melvin “Newman Projection” Newman shows that actylenes can be oxidized to diketones at mildly basic pH (7.5) but will be cleaved to diacids at either strongly acidic or strongly basic pH. - Potassium permanganate oxidations of terminal olefins and acetylenes to carboxylic acids of one less carbon
A. Paul Krapcho, James R. Larson, and Joyce M. Eldridge
The Journal of Organic Chemistry 1977 42 (23), 3749-3753
DOI: 10.1021/jo00443a026
why doesn’t Dihydroxylation With OsO4 work on alkynes ecen though Dihydroxylation With KMnO4 dose work on alkynes
That is a great question. Very simple question, but difficult to answer. Related question: ruthenium is just above Os in the periodic table, but RuO4 will oxidize alkynes to diketones whereas OsO4 does not. Why?
The best I can find is this paper, which covers alkenes (not alkynes). The gist of it is that Os is more stable at higher oxidation states (e.g. Os(VIII), Os(VI) ) relative to smaller elements such as Ru and presumably Mn.
RuO4 is more similar in many respects to KMnO4 than it is to OsO4 – will do many of the same reactions, even though they are not even in the same column of the periodic table.
I suppose the best answer I can give you is that chemistry is as yet an incomplete science, and there are some questions we are still trying to figure out the answers to.
can you a give an intutituve explanation regarding the failiure of dihydroxylation of alkynes via OsO4 sir?
Alkynes are generally less electron-rich than alkenes, owing to the larger electronegativity of sp hybridized carbon atoms. Electrophilic reagents like ozone, HX and OsO4 tend to react more slowly with alkynes.
Regarding why OsO4 doesn’t work, I’m not entirely sure.
http://link.springer.com/article/10.1023%2FA%3A1012733023065#page-1
this paper also show cyclopropanation of an alkyne.
Alkynes CAN be cyclopropanated, it’s just that this topic almost never comes up in introductory organic chemistry.
The comment on OsO4 not working with alkynes prompted me to look it up. It is discussed here on page 10.
http://nptel.ac.in/courses/104103023/download/module1.pdf
When does it work, and when doesn’t it?
That is new to me.
This didn’t appear in my copy of March, nor any other book in my collection, and I don’t have Scifinder at my fingertips anymore to really dig into the literature.
I’d need to see the primary literature to know the exact conditions for the reaction. It’s very possible to selectively dihydroxylate an alkene in the presence of an alkyne; reactions with alkynes are much slower.
Thanks for adding this.
For what it’s worth, I cannot find the first reaction (oxidation of diphenylacetylene to benzil with osmium tetroxide) on Reaxys. Quite a variety of oxidants have been used for this particular substrate, including KMnO4 which is perhaps more familiar to undergrads (JOC 1989 5182), but not OsO4.
The second substrate is even more dubious. There is only one report of this transformation (TL 2004 8575) using trifluoro DMDO, not OsO4, and the authors report that the 1,2-diketone was only a side product, obtained in 18% yield (the main product being that with the hydroxyls oxidised to carbonyls). I don’t know where the “OsO4/KClO3” comes from, but it’s not in the primary literature.
Thanks for digging through the literature, Jon. Much appreciated.