Carboxylic Acid Derivatives
Addition-Elimination Mechanisms With Neutral Nucleophiles (Including Acid Catalysis)
Last updated: May 28th, 2026 |
The Second-Most Important Mechanism In Carbonyl Chemistry – Carbonyl Elimination
- The reverse of nucleophilic addition to the C=O bond (giving a tetrahedral intermediate) is elimination of a leaving group from the tetrahedral intermediate to re-form the C=O bond.
- This is called, “1,2-elimination” or sometimes just “elimination” and is a key mechanism of the carbonyl functional group.
- Confusingly, eliminations of alkyl halides are also called 1,2-eliminations. See The E2 Mechanism or E1cB or the E1 Reaction for more on these.)
- Eliminations are generally favored when expulsion of a leaving group results in formation of a weaker base
- Acid catalysis is extremely helpful in promoting nucleophilic acyl substitution of carboxylic acids and amides, since elimination results in the much better leaving groups H2O and NH3 (as opposed to O2- and (-)NH2 under basic conditions)
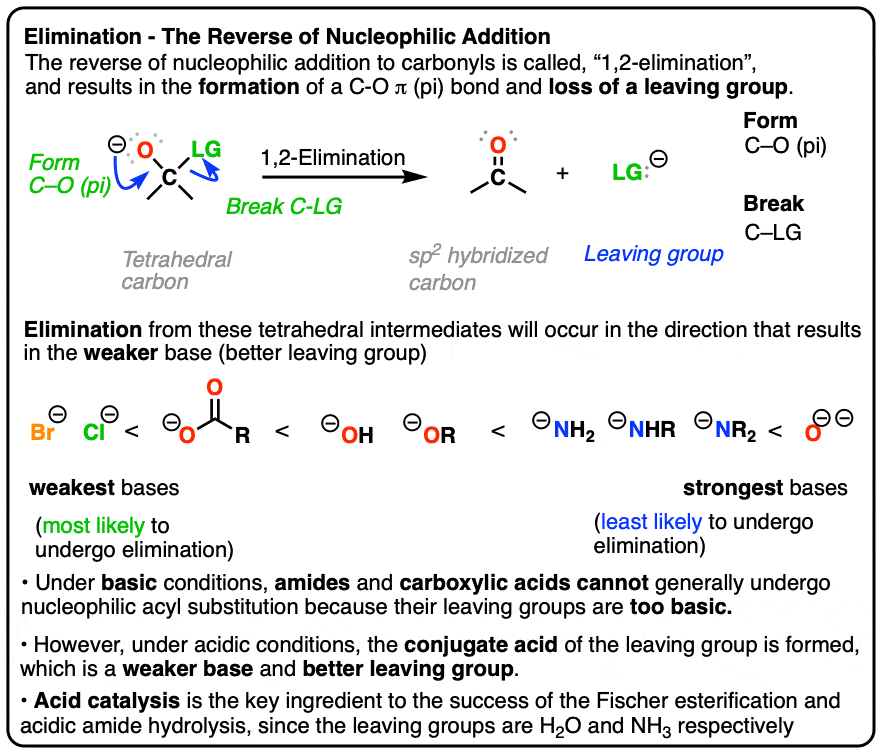
Table of Contents
- Elimination: The Second-Most Important Mechanism of the Carbonyl Group
- Elimination in Carboxylic Acid Derivatives
- What About Neutral Nucleophiles In Nucleophilic Acyl Substitution?
- Acid Catalysis For Nucleophilic Acyl Substitution With Neutral Nucleophiles
- Examples of Acid Catalysis
- Summary
- Notes
- Quiz Yourself!
- (Advanced) References and Further Reading
1. The Second-Most Important Mechanism Of The Carbonyl Group
Elimination (1,2-elimination) is an extremely important reaction mechanism of the carbonyl (C=O) group, which is present in such functional groups as aldehydes, ketones, carboxylic acids and carboxylic acid derivatives.
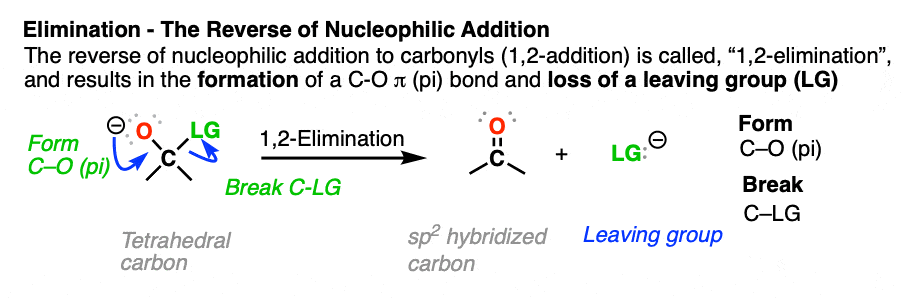
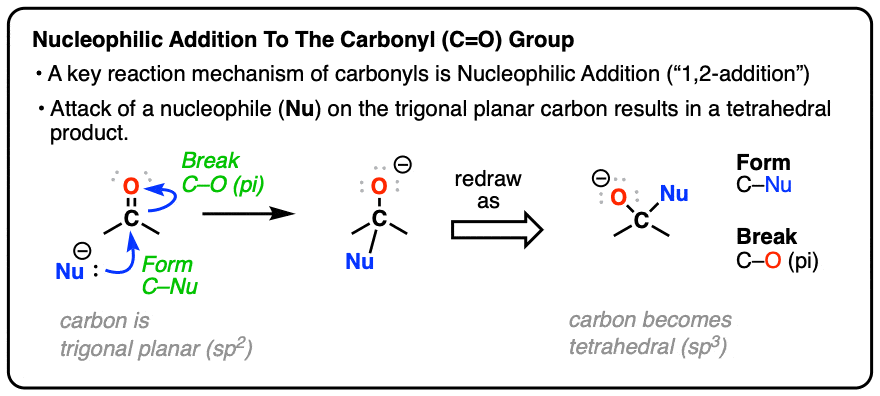
It’s the second most important reaction of carbonyls, after addition. In fact, it is the exact reverse of this nucleophilic addition reaction. (See post: Nucleophilic Addition)
For a reminder of the nucleophilic addition mechanism, hover here or click this link.
{kind=link}
Elimination reactions tend to be favorable when the leaving group is a weaker base than the nucleophile. (See: What Makes A Good Leaving Group)
That’s why halide ions don’t successfully perform nucleophilic addition to aldehydes and ketones. The reaction is going uphill in terms of basicity. Since Cl(-) is a weaker base than O(-), elimination is much more favorable than addition. (See post: How to Use a pKa Table)
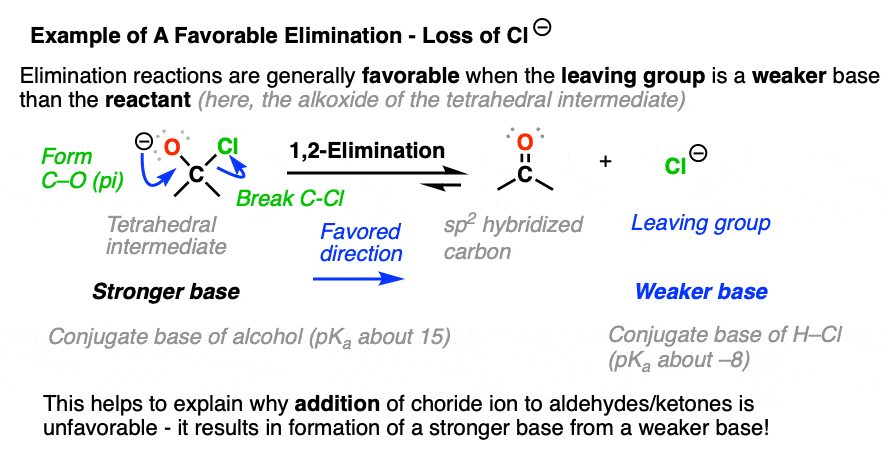
Elimination is unfavorable when it results in a stronger base being formed from a weaker base. This is why reduction of aldehydes and ketones with hydride reducing agents like NaBH4 is irreversible (See: Sodium Borohydride).
The forward reaction for hydride reduction of aldehydes and ketones results in a stronger base (hydride, conjugate base of H2, pKa about 35) being converted to a weaker base (alkoxide, conjugate base of alcohol, pKa about 16-18).
The opposite reaction (elimination) is about 20 pKa units more disfavored from an acid-base perspective (See post: How to Use a pKa Table)
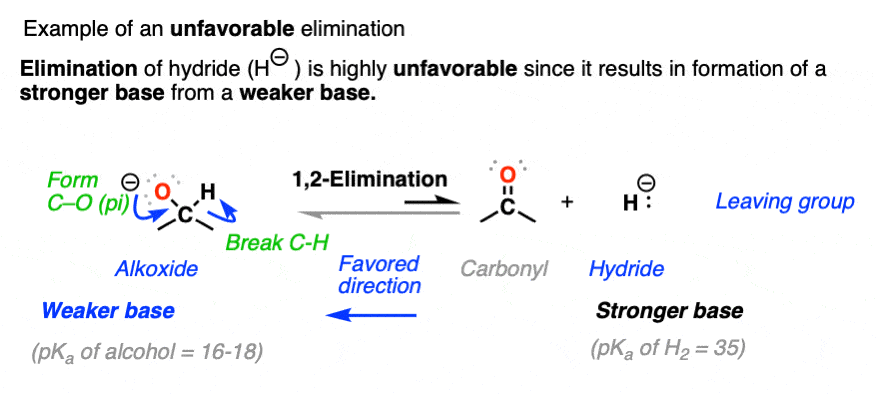
In general, a good rule of thumb is that if the nucleophile/base and leaving group are separated by more than 8 pKa units, the reaction can be considered to be irreversible. (See: A Handy Rule of Thumb For Acid-Base Reactions)
(However, there are some examples of reactions where H(-) can act as a leaving group in a concerted process – See Note 1)
The Principle of Acid-Base Mediocrity (“stronger acid plus stronger base gives weaker acid plus weaker base) keeps coming up again and again!
2. Elimination In Carboxylic Acid Derivatives
Besides aldehydes and ketones, addition and elimination also occur in the reactions of carboxylic acid derivatives such as acid halides, acid anhydrides, esters, and amides.
When nucleophilic addition occurs to a carboxylic acid derivative, it forms a tetrahedral intermediate with two potential leaving groups. [ but not the O- Note 2]
- The first potential leaving group is the original nucleophile; elimination of this from the tetrahedral intermediate would just give us back our starting material (after all, leaving groups are really just nucleophiles acting in reverse).
- The other potential leaving group is the X of the carboxylic acid derivative (e.g. (–)Cl for acid chlorides, (–)OCOR for anhydrides, (–)OR for esters, (–)NR2 , (–NHR), –(NH2) for amides)
When the nucleophile is a stronger base than the leaving group X(-) of the carboxylic acid derivative, then we will end up swapping out the X group of the carboxylic acid derivative. This is nucleophilic acyl substitution. (See post: Nucleophilic Acyl Substitution With Anionic Nucleophiles)
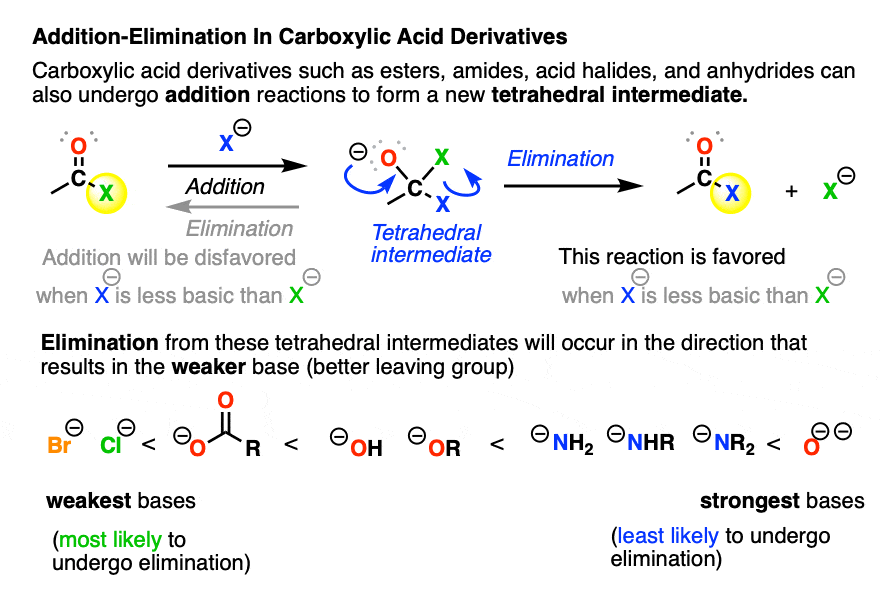
With negatively charged nucleophiles, nucleophilic acyl substitution is easy to perform on acid halides and acid anhydrides, more difficult to perform on esters, and essentially impossible to achieve with amides, since that would require loss of the very basic leaving group NH2(-), conjugate base of an amine (pKa 35-38)
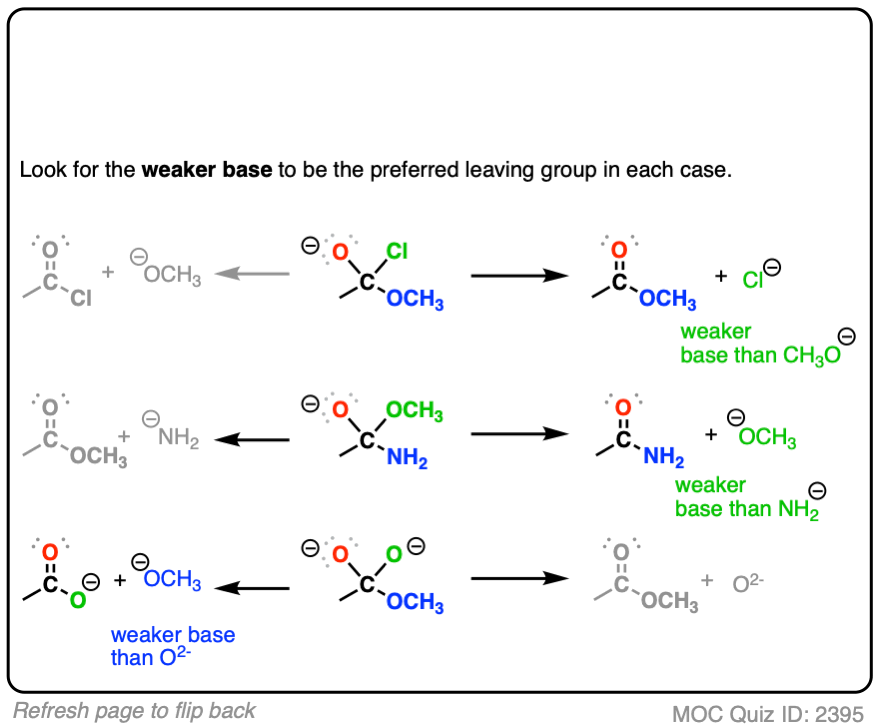
 Click to Flip
Click to Flip
Carboxylic acids generally don’t undergo addition-elimination under basic conditions since they will be deprotonated by strong base to give carboxylates, and the resulting leaving group would have to be the very strong base O(2-) (covered in more detail in this post: Transesterification)
3. What About Neutral Nucleophiles?
It’s not absolutely required to use basic nucleophiles for nucleophilic aromatic substitution, however.
Neutral nucleophiles are perfectly capable of performing some nucleophilic acyl substitutions.
One prominent case is that of acid halides and anhydrides, which are very sensitive to the presence of water.
Both of these functional groups can be hydrolyzed with water to give carboxylic acids.
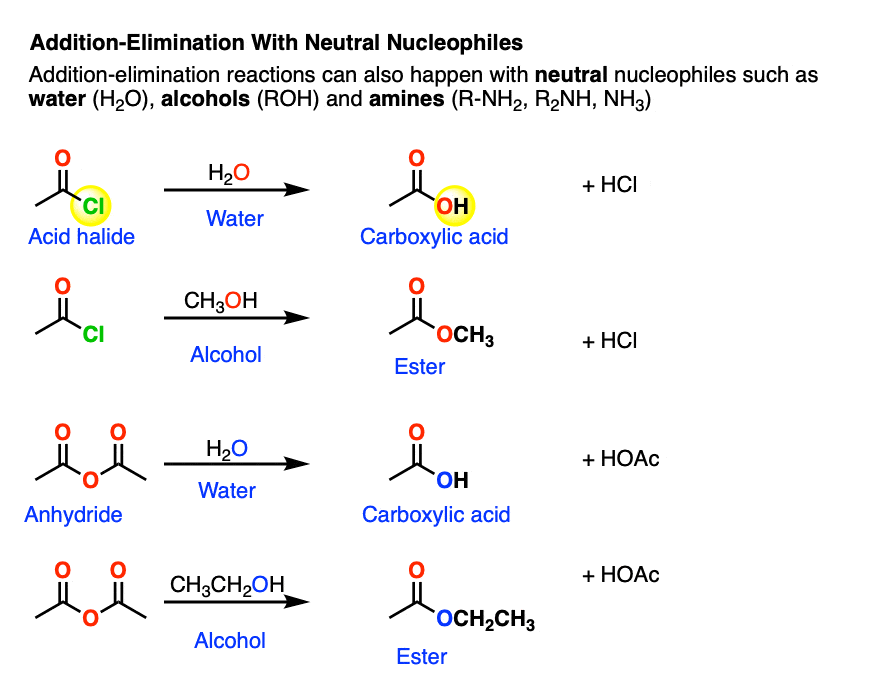
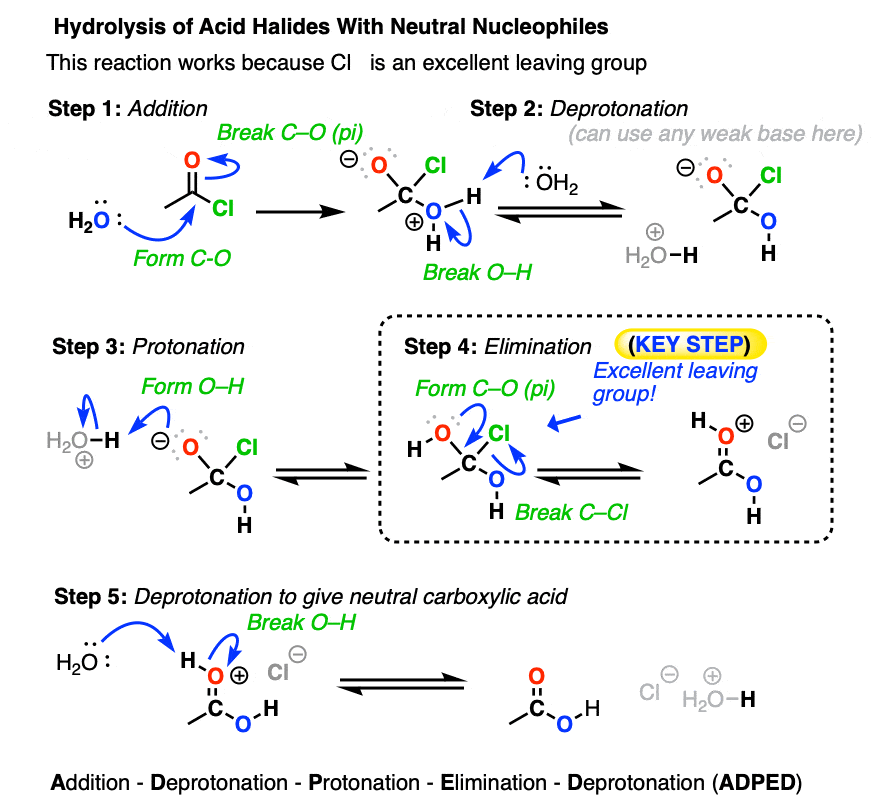
After addition and proton transfer, the key step is elimination of the halide ion, resulting in formation of the carboxylic acid. This still follows the Principle of Acid-Base Mediocrity, since Cl(-) is a weaker base than H2O (conjugate base of H3O+, pKa 0) . Likewise, water is at least comparable in base strength to carboxylic acids.
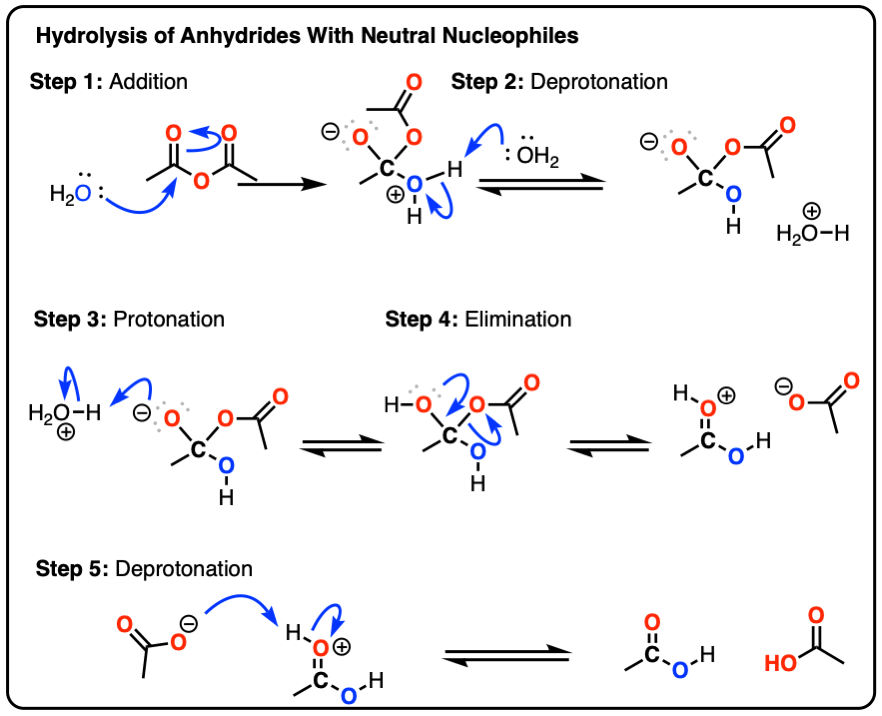
To see a mechanism for hydrolysis of acid anhydrides with water, hover here or click this link.
{kind=link}
These reactions also work well with amines. The reactions between acid halides and amines are some of the best ways for making amides; this reaction is sometimes known as the Schotten-Baumann reaction (See Synthesis of Amides From Acid Halides).
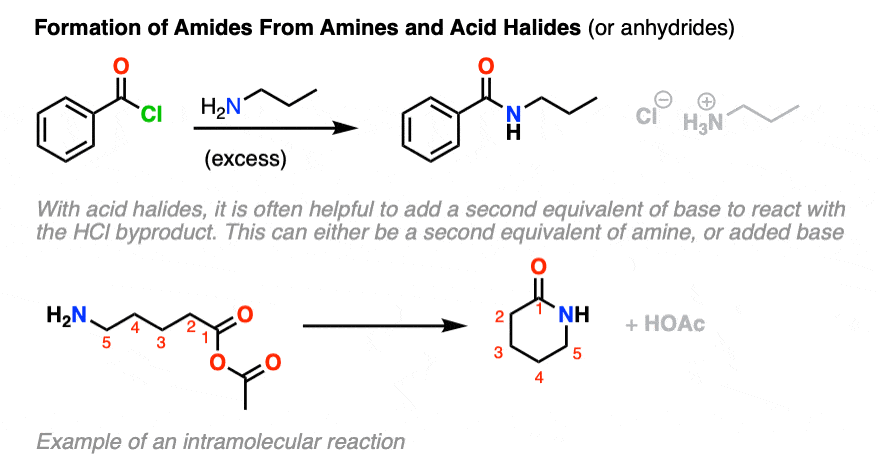
Generally, at least two equivalents of amine are used, since one equivalent of HCl is generated. (If only one equivalent of amine were used, the reaction would not proceed to completion since the amine nucleophile would be protonated to give (non-nucleophilic) RNH3(+). )
To see a mechanism for the formation of amides from acid halides, hover here or click this link.
{kind=link}
4. Acidic Conditions – Elimination Of The Conjugate Acid (A Better Leaving Group)
Addition of neutral nucleophiles to carboxylic acids has its limits, however. Water can displace weakly basic halides and carboxylates, but can’t displace the much stronger bases RO(-) or R2N(-). (Amines, if heated might displace esters, but it requires a lot of heat – See Synthesis of Amides).
Aside from acid halides and anhydrides, most other carboxylic acids are inert under neutral conditions.
However, when an acid catalyst is added, it’s a different story altogether. It opens up a whole different set of reactions that don’t happen otherwise.
We’ve previously seen an example of this in the synthesis of acetals (See post: Acetals and Hemiacetals) where elimination of one equivalent of ROH from a hemiacetal required an acid catalyst. (In the absence of acid, no elimination happens!).
For a reminder of the elimination mechanism in acetal synthesis, hover here or click this link.
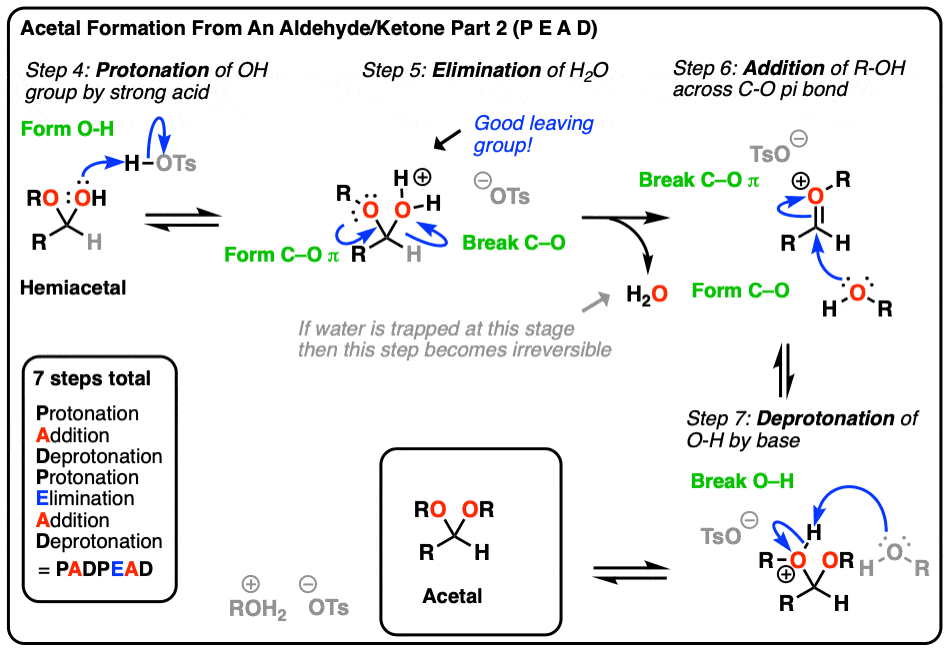{kind=link}
This is also the case for carboxylic acid derivatives.
Recall that the whole reason amides and carboxylic acids are unreactive under basic and neutral conditions is because NH2(-) and O(-) are such strong bases and therefore poor leaving groups.
But just imagine for a moment that we could run these reactions under acidic conditions.
Their leaving groups would no longer be NH2(–) and O(2-) ; they could be their protonated cousins NH3 and H2O , which are much weaker bases and therefore much better leaving groups.
Suddenly, substitution reactions of carboxylic acids and esters becomes a plausible reaction!
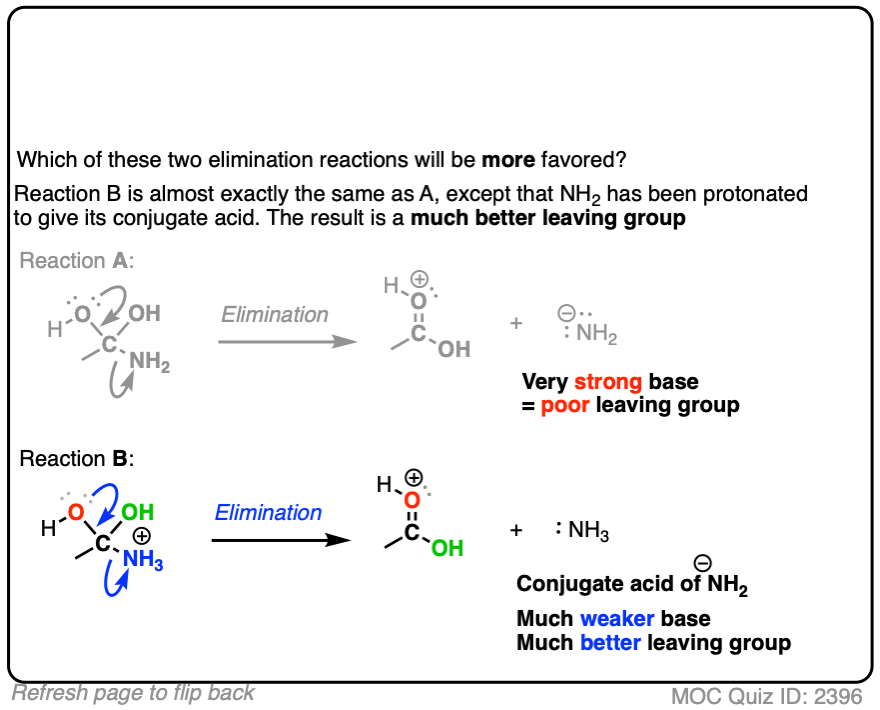
Compare the leaving groups in these two reactions
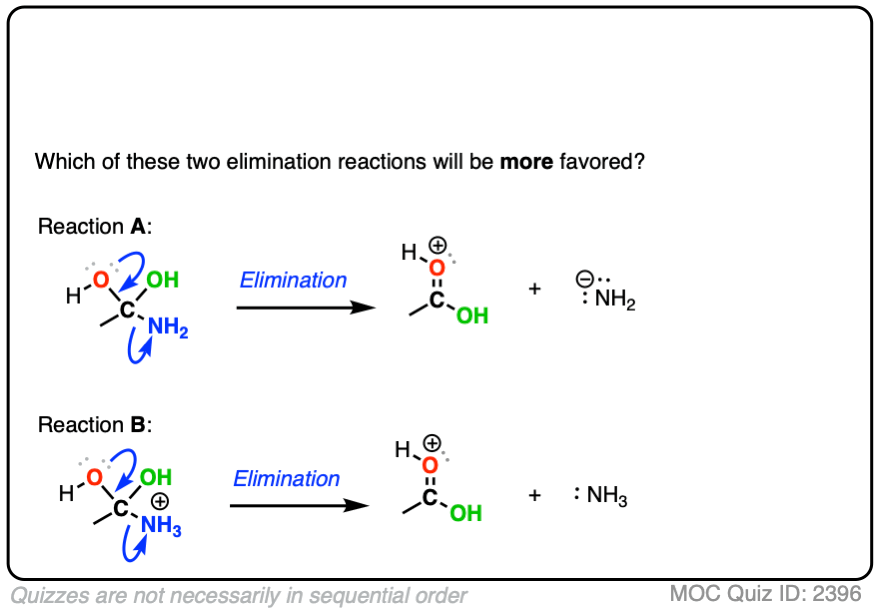 Click to Flip
Click to Flip
Or the leaving groups in these two reactions
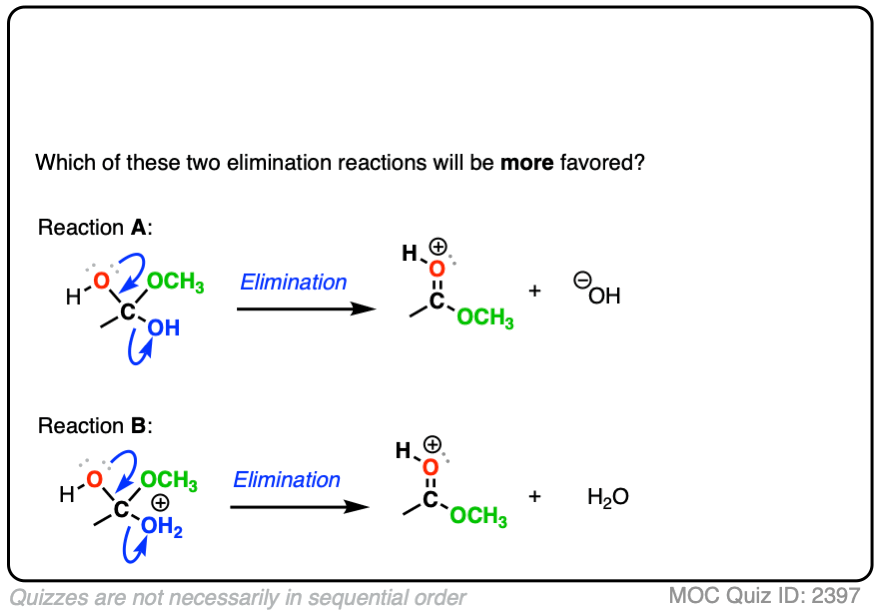 Click to Flip
Click to Flip
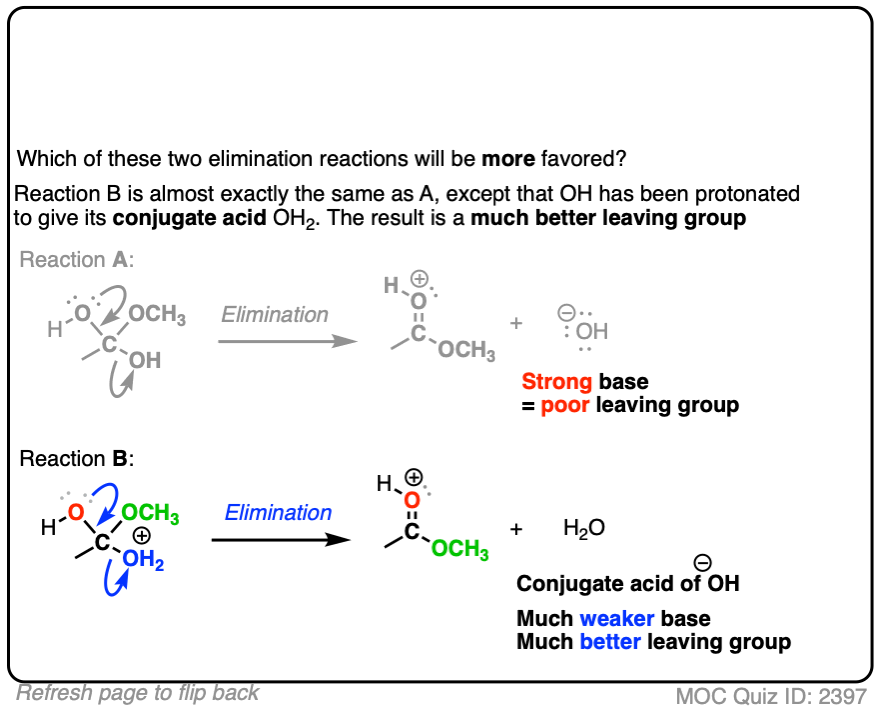
In each case, the protonated species is the better leaving group, because it is the weaker base.
The conjugate acid is a better leaving group, which is one important reason why acid catalysis helps with elimination reactions.
5. Examples Of Acid Catalysis In Nucleophilic Acyl Substitution
Here are two examples of using acid as a catalyst in nucleophilic acyl substitution reactions, and how acid assists with loss of a leaving group.
A carboxylic acid can be converted into an ester under acidic conditions. This is the Fischer Esterification (See post: Fischer Esterification).
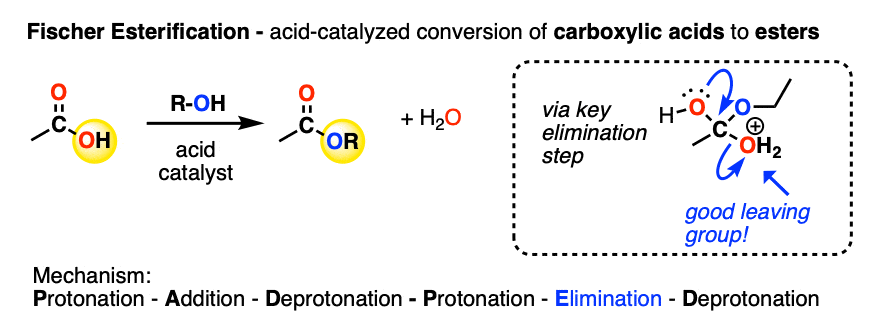
In the key step, a molecule of H2O is eliminated from the tetrahedral intermediate, resulting in a protonated ester.
Similarly, amides can be converted to a carboxylic acid under acidic conditions. This is called acidic amide hydrolysis (See post: Amide Hydrolysis)
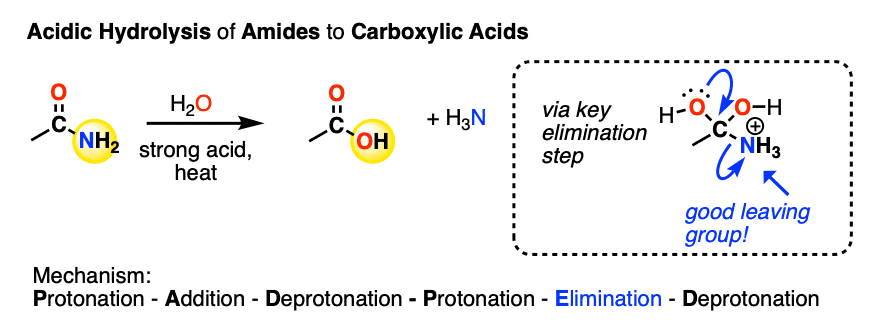
Likewise, the key step is elimination of the weak base NH3 from the tetrahedral intermediate. In the absence of acid, no substitution occurs.
One limitation of acid catalysis for elimination is that we’re limited to using nucleophiles that aren’t irreversibly destroyed by strong acid.
This means that strongly basic nucleophiles like NH2(-), HO(-), RO(-), hydrides and Grignards are incompatible with acidic conditions, since acid-bases reactions are generally much faster than any reactions at carbon. (See post: Acid-Base Reactions are Fast),
6. Summary – Conclusion
- If nucleophilic addition is the most important reaction mechanism of carbonyls, then its opposite – elimination – ranks as the second-most important mechanism.
- It’s a key mechanism in nucleophilic acyl substitution reactions, in addition to its role in formation of acetals and imines that we saw previously in the chapter on aldehydes and ketones.
- The favorability of elimination is determined by the basicity of the leaving group. Good leaving groups are weak bases.
- Poor leaving groups can be transformed into good leaving groups through the addition of acid, since the conjugate acid of any species is a weaker base and therefore a better leaving group.
- Addition of acid is essential in the nucleophilic substitution reactions of carboxylic acids and amides such as the Fischer esterification and acidic amide hydrolysis.
Notes
Note 1. One example where hydride does leave in an elimination-type process is in the Canizarro reaction of aldehydes. The key step is transfer of a hydride from a (deprotonated) aldehyde hemiacetal to a second equivalent of the same aldehyde, resulting in an alkoxide and a carboxylic acid (which quickly undergo an acid-base reaction).
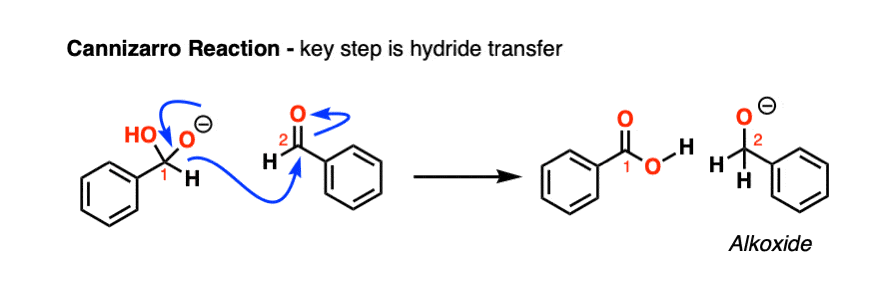
However, this reaction requires extremely basic conditions and heat (as well as requiring non-enolizable aldehydes)
Note 2. But not O- , since this would have to leave as O2-
Quiz Yourself!
Become a MOC member to see the clickable quiz with answers on the back.

Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading
- SN2 Mechanism for Alcoholysis, Aminolysis, and Hydrolysis of Acetyl Chloride
T. William Bentley, Gareth Llewellyn, and J. Anthony McAlister
The Journal of Organic Chemistry 1996 61 (22), 7927-7932
DOI: 10.1021/jo9609844
Although addition-elimination reactions are assumed in most cases, there are situations where nucleophilic acyl substitution results from direct attack at a carbonyl carbon with a nucleophile, especially in highly polar solvents where the leaving group is easily ionized. - Computational Studies of Nucleophilic Substitution at Carbonyl Carbon: the SN2 Mechanism versus the Tetrahedral Intermediate in Organic Synthesis
Joseph M. Fox, Olga Dmitrenko, Lian-an Liao, and Robert D. Bach
The Journal of Organic Chemistry 2004 69 (21), 7317-7328
DOI: 10.1021/jo049494z - Estimates of hydride ion stability in condensed systems: energy of formation and solvation in aqueous and polar-organic solvents
- BENZOYL PIPERIDINE
Marvel, C. S.; Lazier, W. A.
Org. Synth. 1929, 9, 16
DOI: 10.15227/orgsyn.009.0016
This procedure from Organic Syntheses, a source of independently tested and reproducible synthetic organic laboratory procedures, is a classic Schotten-Baumann amide synthesis.The original Schotten-Baumann papers: - Ueber die Oxydation des Piperidins
Schotten, C.
Ber. 1884, 17 (2), 2544-2547
DOI: 10.1002/cber.188401702178 - Ueber eine einfache Methode der Darstellung von Benzoësäureäthern
Baumann, E.
Ber. 1886, 19 (2), 3218-3222
DOI: 10.1002/cber.188601902348
When aliphatic primary amines react with esters, for example, butylene diamine with dimethyl carbonate, the reaction rate is quite slow, but when water is added, it become very quick. How to explain this phenomenon?
Dimethyl carbonate is not an ester, it is a carbonate, which is a different functional group.
One of the key reactions of amines with carbonates is to install protective groups, such as the Boc group, and it is generally done under basic conditions (i.e. not just water, but water in the presence of a base such as sodium carbonate). Representative procedures can be found on this page:
https://www.organic-chemistry.org/protectivegroups/amino/boc-amino.htm
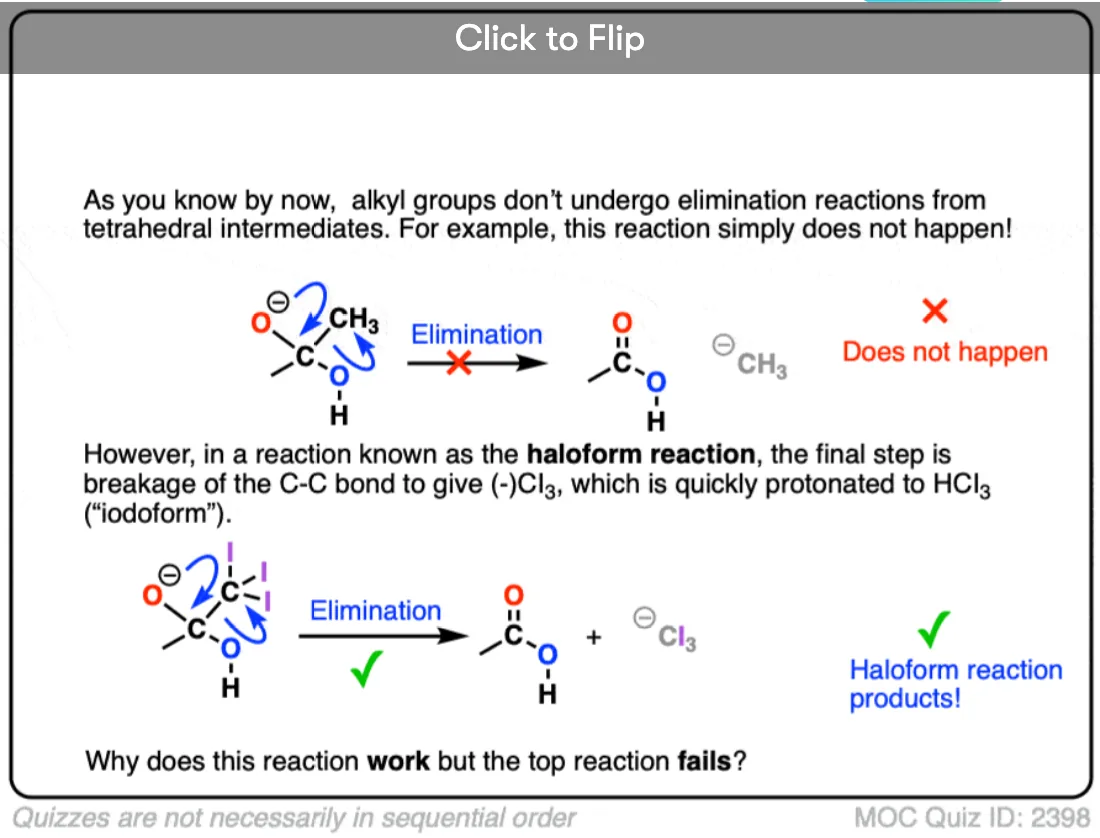
Would the haloform reaction which involves nucleophilic attack by OH followed by displacement of a CBr3 group also count?
Yes – great example. The Cl3C(-) anion leaves via a [1,2] elimination reaction.
“…(Halides, AcO(-),…”
What does AcO(-) mean?
Thanks!
AcO(-) means acetyl: CH3C(O)O- , the conjugate base of acetic acid