Dienes and MO Theory
Why Are Endo vs Exo Products Favored in the Diels-Alder Reaction?
Last updated: May 28th, 2026 |
[Advanced] Secondary Orbital Interactions – A Rationale For Why Endo Products Are Favored In The Diels-Alder Reaction
In our last post, we noted that endo products tend to be favored over exo products in the Diels-Alder reaction. [We also introduced a quick-and-dirty rule for telling the difference between endo- and exo- products in the Diels-Alder.]
So why are endo products generally favored over exo in the Diels-Alder reaction? That’s what we’re going to learn about today…
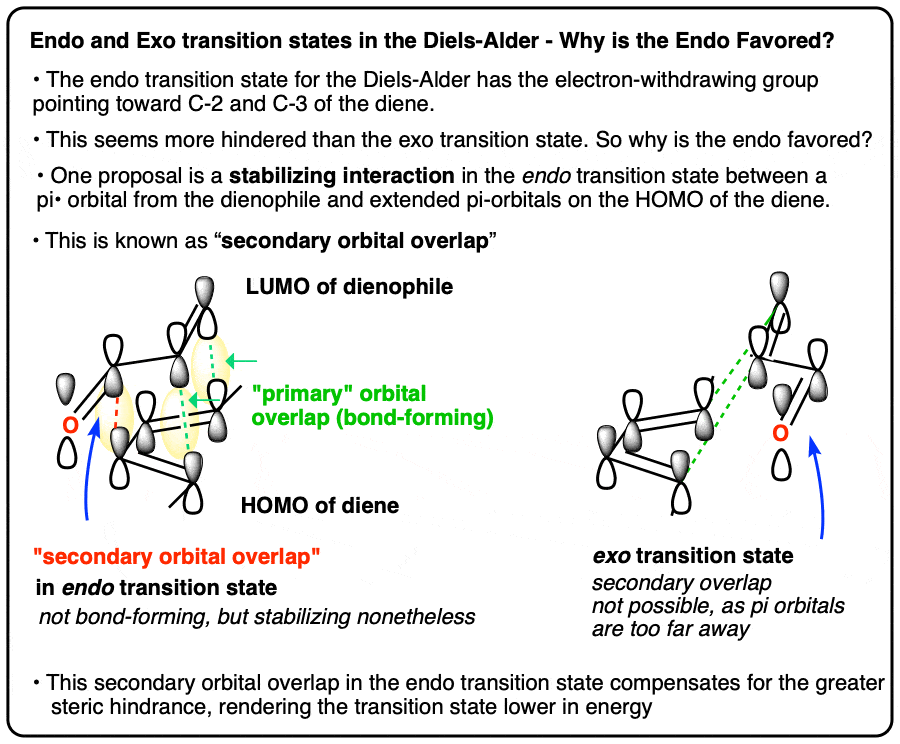
Table of Contents
- Endo Products Tend To Be Favored In The Diels-Alder Even Though They Are More Sterically Hindered
- The Endo And Exo Transition States For The Diels-Alder Reaction Between Cyclopentadiene And Maleic Anhydride
- Another Pair Of Endo And Exo Diels-Alder Transition States Shows That The Endo Transition State Places The EWG On The Dienophile Over The Diene
- Secondary Orbital Interactions Are Possible In The Endo Transition State (But Not The Exo)
- Kinetic and Thermodynamic Control of the Diels Alder Reaction
- Notes
- Quiz Yourself!
- (Advanced) References and Further Reading
1. Endo Products Tend To Be Favored In The Diels-Alder Even Though They Are More Sterically Hindered
The preference for endo versus exo is especially curious since the endo products appear to be more sterically hindered.
For example, here’s the Diels-Alder reaction of cyclopentadiene with maleic anhydride. The ratio of endo to exo products in this reaction is about four to one:
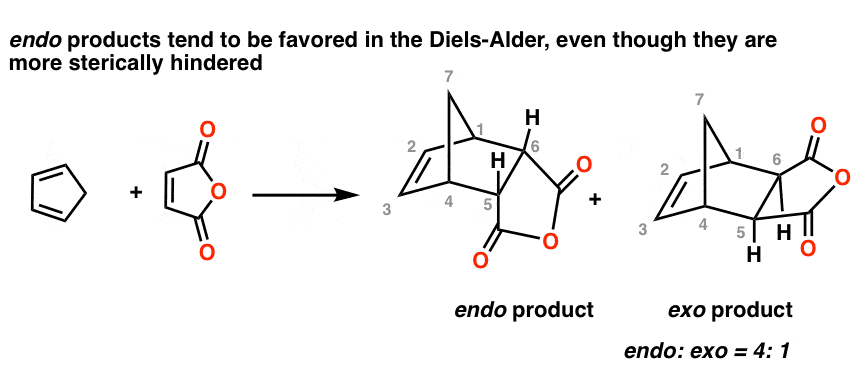
Note that in the endo product above, the anhydride is on the underside of the new six-membered ring, whereas in the exo, it points away. This is indeed less sterically hindered.
[To jump ahead, here’s a fact we’ll cover in more detail in the next post: most exo products are in fact more stable than the endo products for steric reasons, but the endo product tends to be formed faster. Furthermore, given enough heat, the Diels-Alder product can revert back to starting materials . Long story short: the Diels-Alder is another example of a reaction that can be run under kinetic or thermodynamic control, where the “endo” is the kinetic product and the “exo” is the thermodynamic product. ]
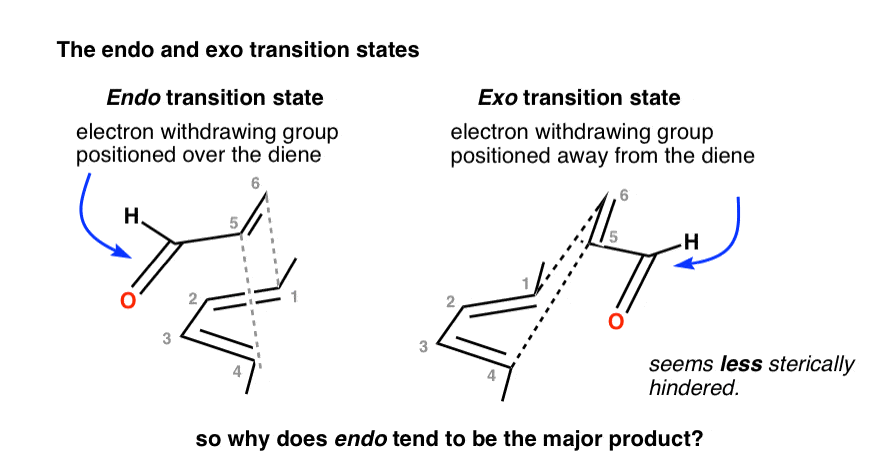
2. The Endo And Exo Transition States For The Diels-Alder Reaction Between Cyclopentadiene And Maleic Anhydride
So why is the endo typically favored?
Here’s one attempt at an explanation, using “secondary orbital effects” [Note 1]. Another related explanation is in the footnotes. [Note 2].
Our last post showed the mechanism of the Diels-Alder reaction through the orbital interactions of the highest occupied molecular orbital (HOMO) of the diene and the lowest unoccupied molecular orbital (LUMO) of the dienophile.
When the orbital interactions are drawn out in the two different transition states, we see something like this.
Note that in the endo transition state the electron-withdrawing groups point toward the two carbons that will eventually comprise the C2-C3 alkene, whereas in the exo transition state, the electron-withdrawing groups point away from the C2-C3 alkene:
3. Another Pair Of Endo And Exo Diels-Alder Transition States Shows That The Endo Transition State Places The EWG On The Dienophile Over The Diene
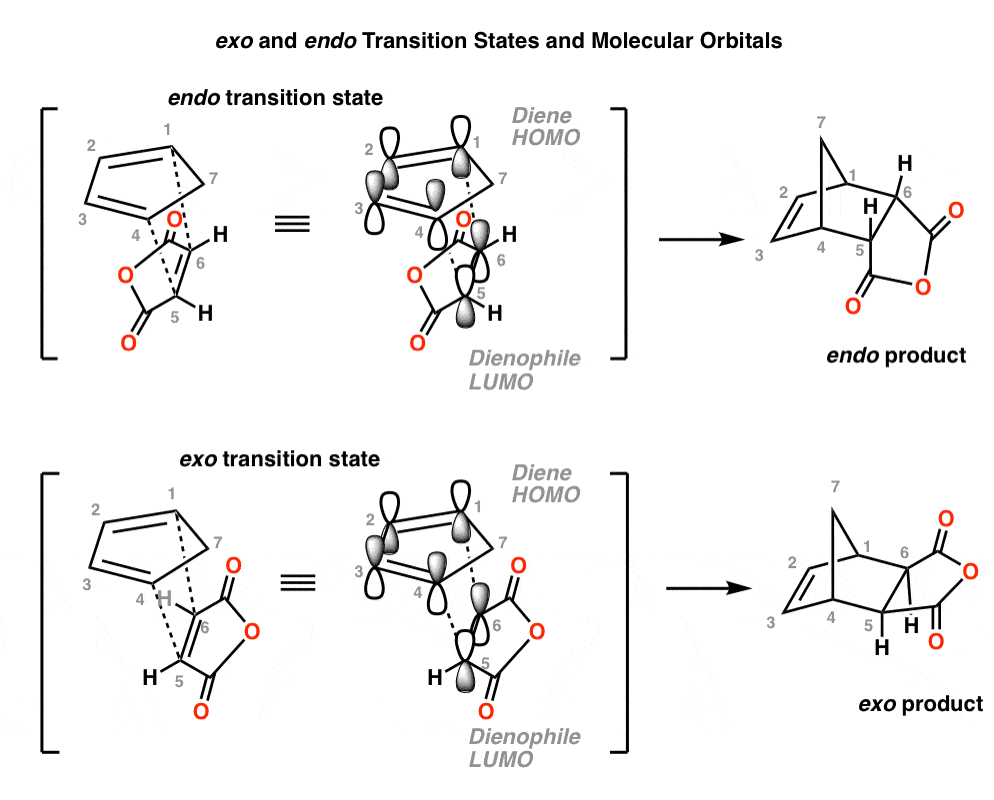
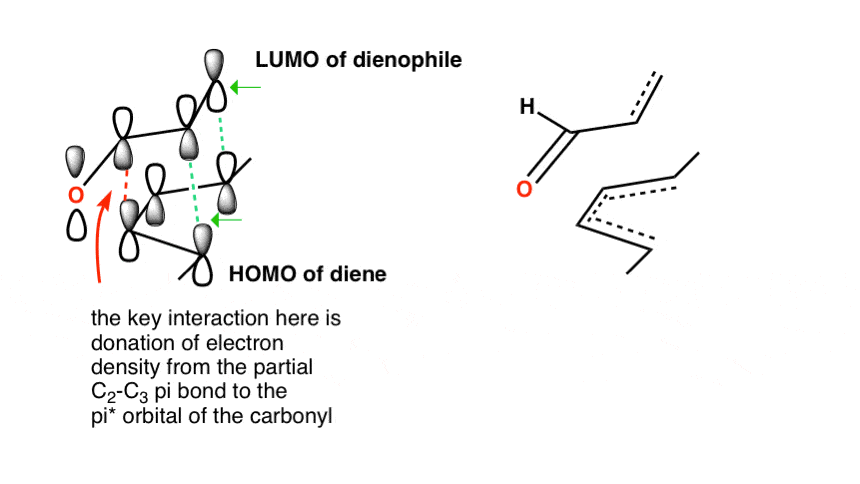
Here’s another example with (E,E)-2,4-hexadiene and acrolein. Again, note how the endo transition state has the electron withdrawing group positioned over the diene:
- In the endo transition state, the carbons which become the C2–C3 double bond are positioned right below the carbonyl carbon of the dienophile.
- In the exo transition state, the electron-withdrawing group points away from the C2–C3 carbons.
4. Secondary Orbital Interactions Are Possible In The Endo Transition State (But Not The Exo)
So what?
By itself, this doesn’t seem to offer any explanation as to why the endo transition state might be favored.
IF we just confine ourselves to examining the “primary” molecular orbitals – i.e. the molecular orbitals involved in bond formation.
The key difference comes when we extend our view and look the “secondary” molecular orbitals of the diene and dienophile that are not directly involved in bond formation, but might still interact with each other.
In this view, there is something special about the endo transition state that isn’t present in the exo.
Because the C2-C3 orbitals of the diene HOMO are positioned close to the C=O orbitals of the dienophile LUMO, they can interact. This isn’t possible in the exo transition state.
This is not a bond-forming interaction (that would be a “primary orbital interaction”), but it is a stabilizing interaction nonetheless. We call it a “secondary orbital interaction”. [We haven’t written about “hyperconjugation” here, but you can think of the interaction as being similar. Essentially, it’s an interaction between an occupied orbital with an unoccupied orbital to form what you can think of as a very weak “partial bond”, and the overall interaction is stabilizing].
This interaction stabilizes the endo transition state to an extent that compensates for the slightly greater steric hindrance.
Here’s what these orbitals look like:
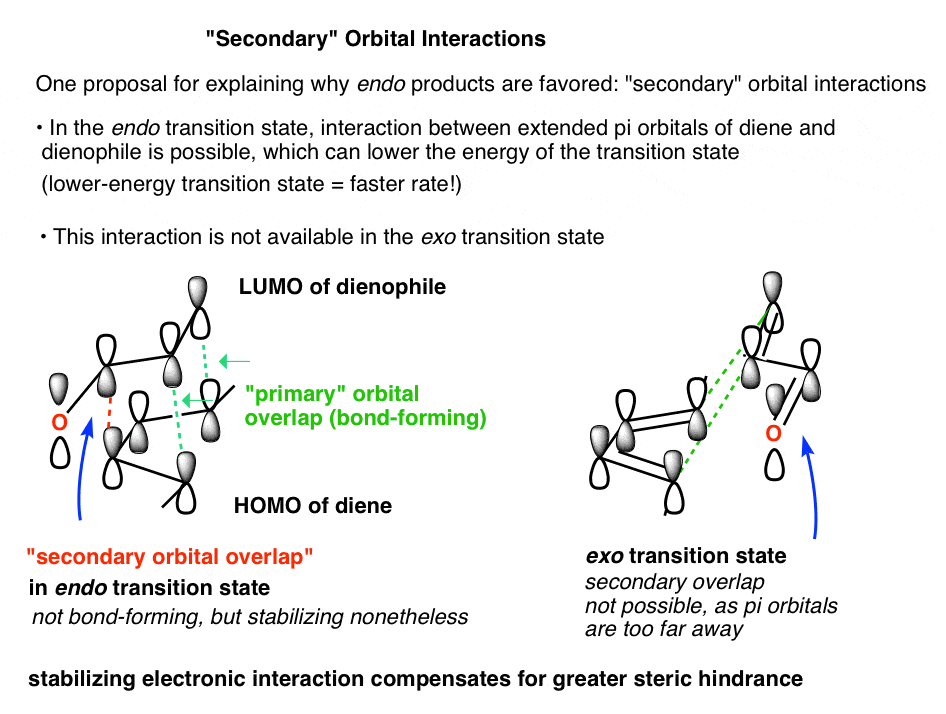
That’s really it. If you know how to draw the complete HOMO of the diene and the complete LUMO of the dienophile, then you can sketch out how they might interact. [We described how to draw out pi molecular orbitals in this post].
We’ve drawn some molecular orbitals for this reaction in the endnotes.
5. Kinetic and Thermodynamic Control of the Diels Alder Reaction
As alluded to above, the endo product tends to be the “kinetic” product, that is, the one that is formed the fastest. Under typical reaction conditions at relatively low temperature, the product distribution reflects the difference in energy between the exo and endo transition states – which is not necessarily the same thing as the difference in energy between the products!
If heated sufficiently, Diels-Alder products can revert to their starting materials, and an equilibrium between the reactants and products can be established. Under these conditions, the product distribution will reflect the difference in energy between the exo and endo products (which tends to favor the exo. )
We’ll go into more detail on the reversibility of the Diels-Alder (and kinetic vs. thermodynamic control) in a future post.
Next Post: Kinetic And Thermodynamic Control In The Diels-Alder Reaction
Notes
In fairness, there is some debate as to whether secondary orbital effects actually exist.
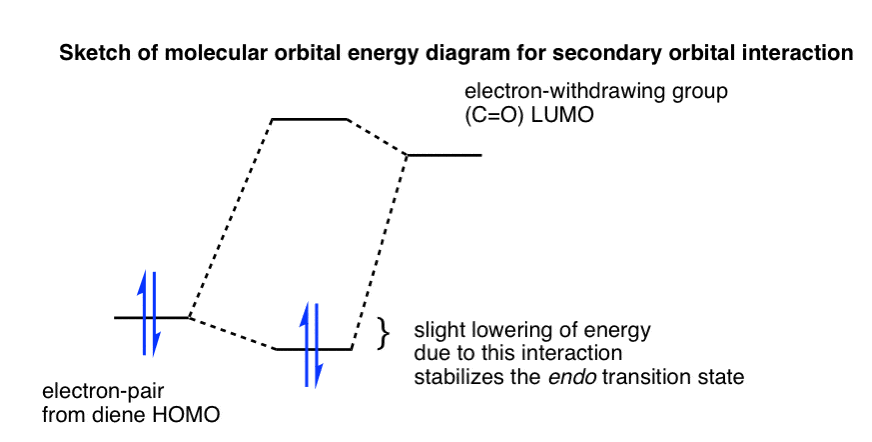
Note 1. Molecular Orbital Diagram for Secondary Orbital Interactions
Here’s another way to look at the preference for endo over exo, which incorporates the same type of interaction. The secondary orbital interaction is marked with a red dashed line, below left.
In the endo transition state, we can have a donation of electron density from electrons in the HOMO of the diene to the empty C=O pi* orbital in the LUMO of the dienophile. This isn’t possible in the exo transition state.
From a molecular orbital perspective, we can draw a pair of electrons in the diene HOMO on the left, and the C=O LUMO on the right.
Any time there is donation from an occupied orbital to an unoccupied orbital there is a lowering of energy.
If the two orbitals in the transition state interact, we can imagine a slight lowering of energy of the electrons (along with a corresponding raise in the energy of the LUMO).
This slight lowering of energy is responsible for the slight preference for the endo transition state.
Lewis Acid Catalysis Increases endo : exo Selectivity
Here’s an extension of the same idea. It’s known that Lewis acids (such as ZnCl2, TiCl4, SnCl4, and many others) can accelerate the rate of Diels-Alder reactions. It can also increase the endo : exo selectivity!
For instance, compare the rate of non-Lewis acid catalyzed versus Lewis-acid catalyzed ratios in this reaction. [Reference]
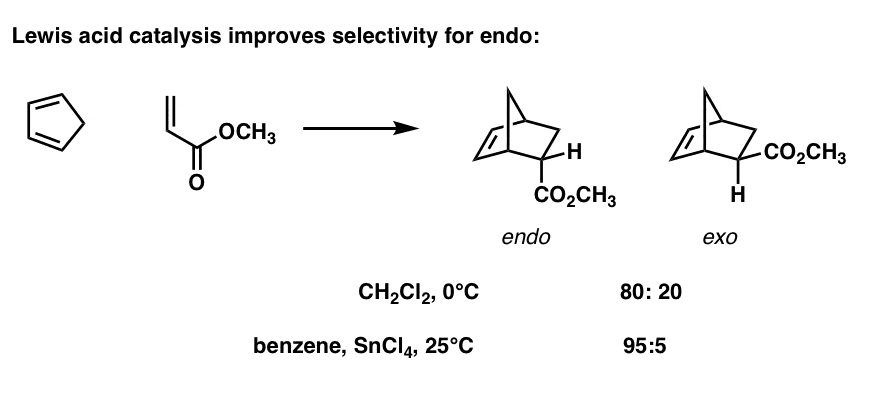
What happens is that the LUMO of the C=O bond is lowered when a Lewis acid coordinates to the carbonyl.
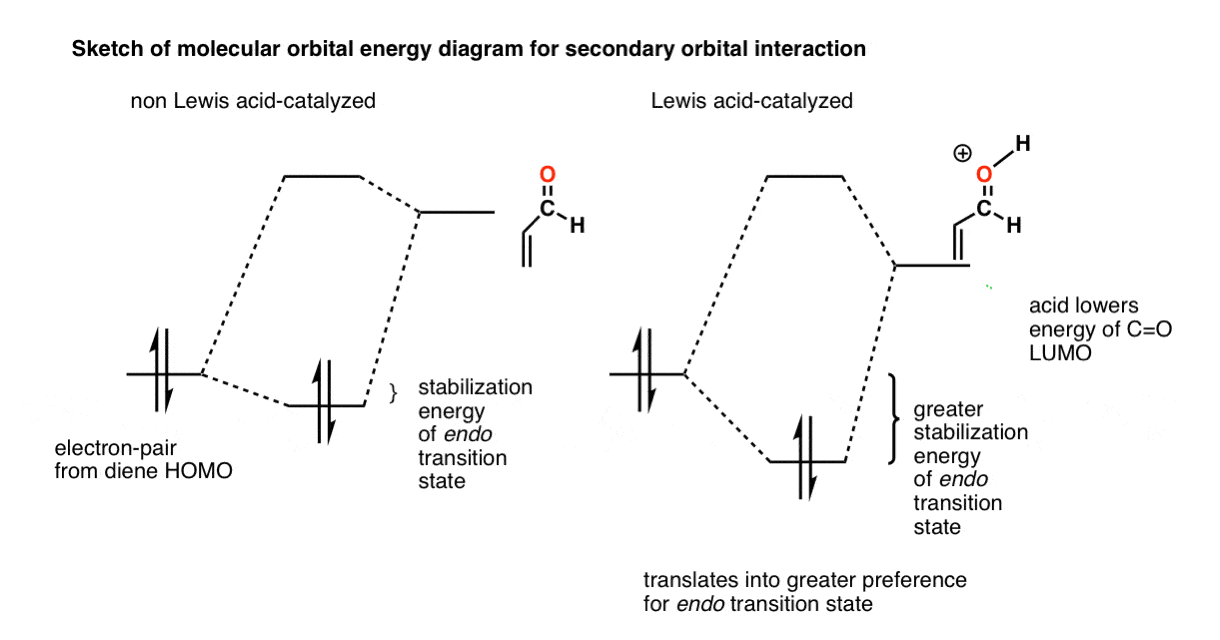
For reasons we won’t explain, the interaction between orbitals strengthens as they become closer together in energy. Therefore the secondary orbital interaction is strengthened and a greater stabilization energy is obtained for the endo transition state.
That’s probably too much for this site, but there you go.
For more information, I highly suggest hunting down Dave Evans’ Chemistry 206 notes from Harvard, if you can find them. They are excellent.
Quiz Yourself!
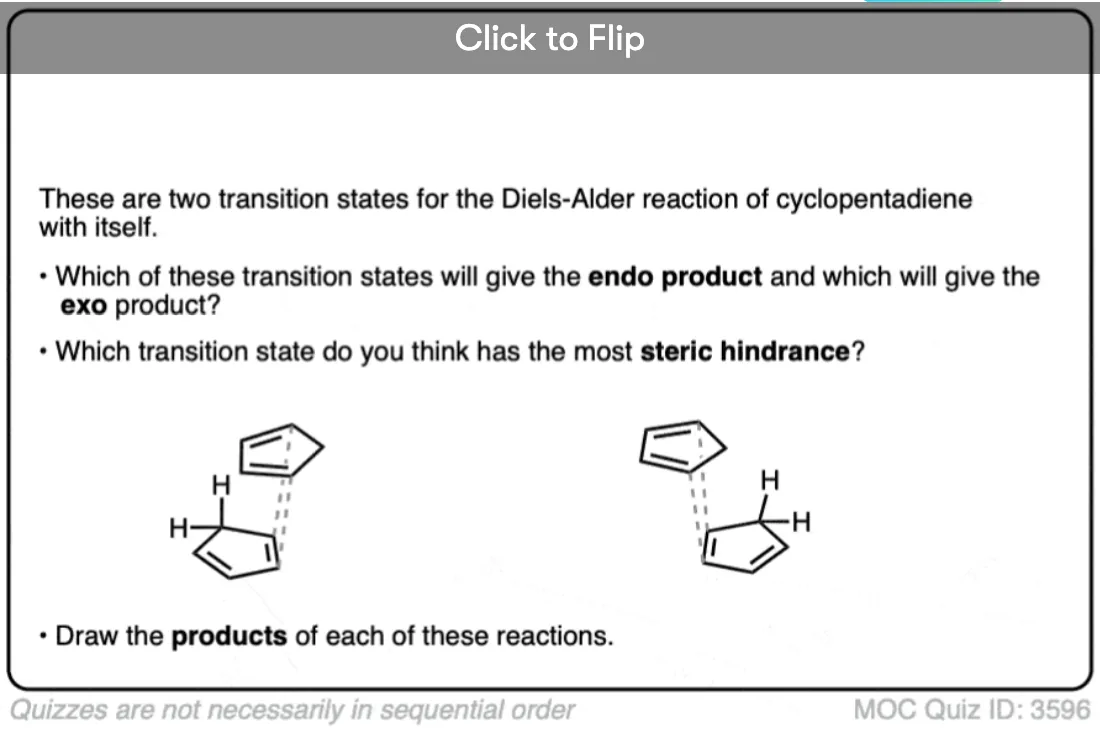
Become a MOC member to see the clickable quiz with answers on the back.
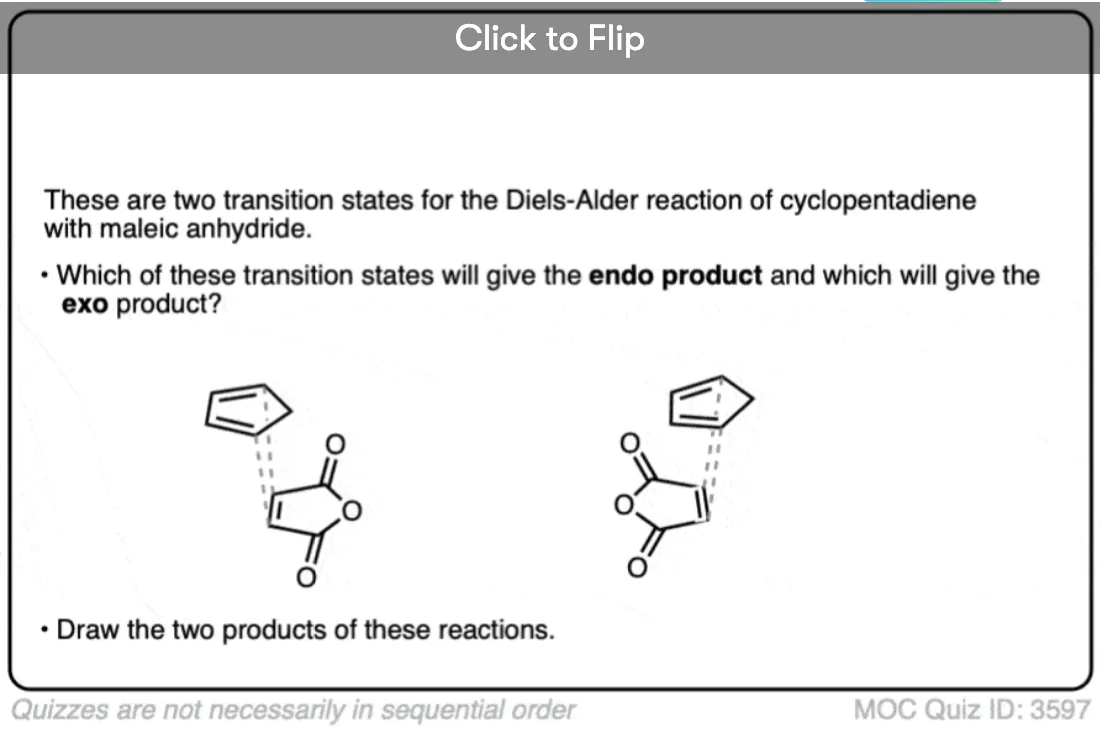
Become a MOC member to see the clickable quiz with answers on the back.
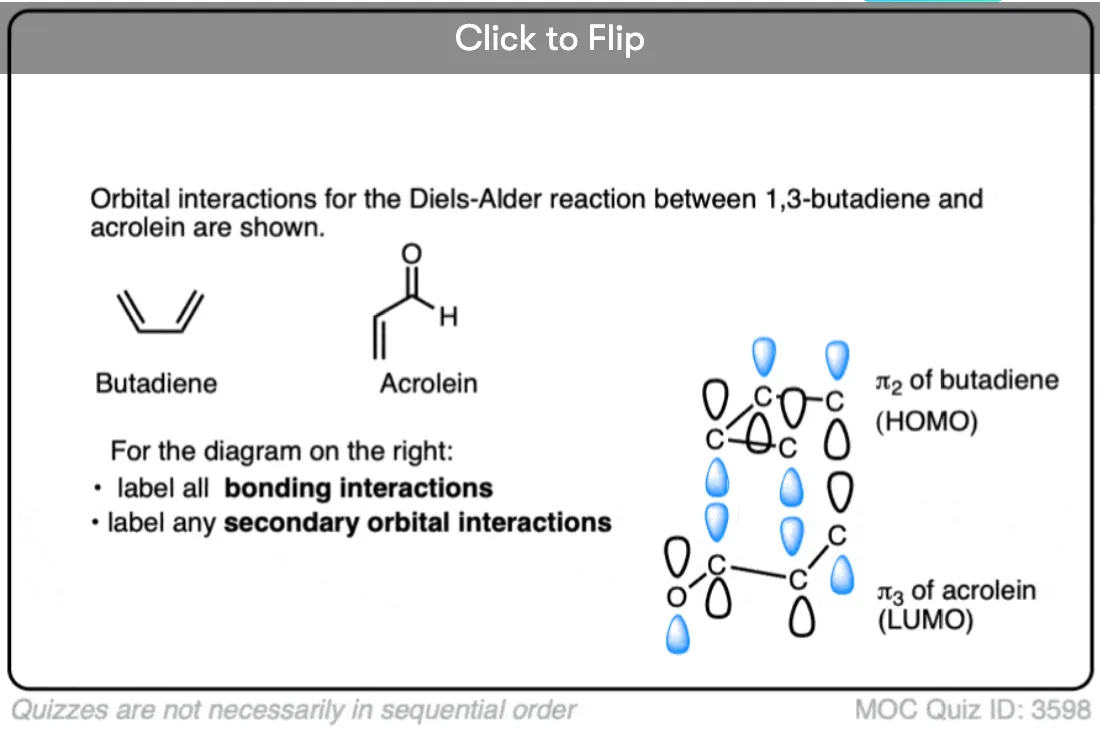
Become a MOC member to see the clickable quiz with answers on the back.
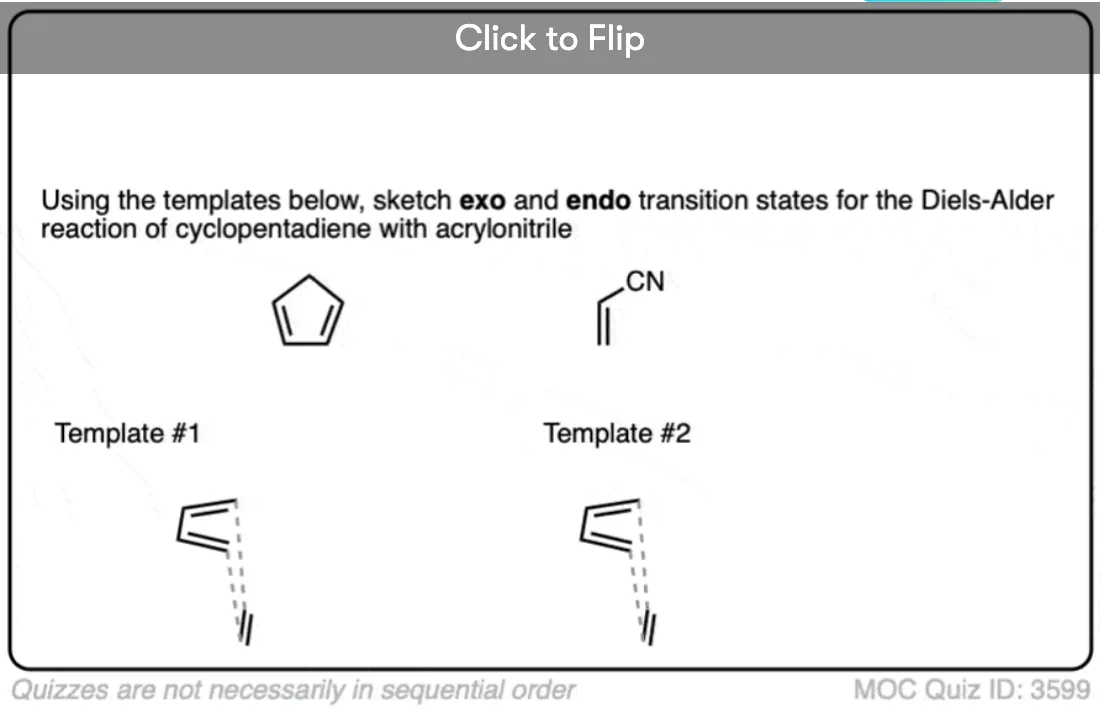
Become a MOC member to see the clickable quiz with answers on the back.
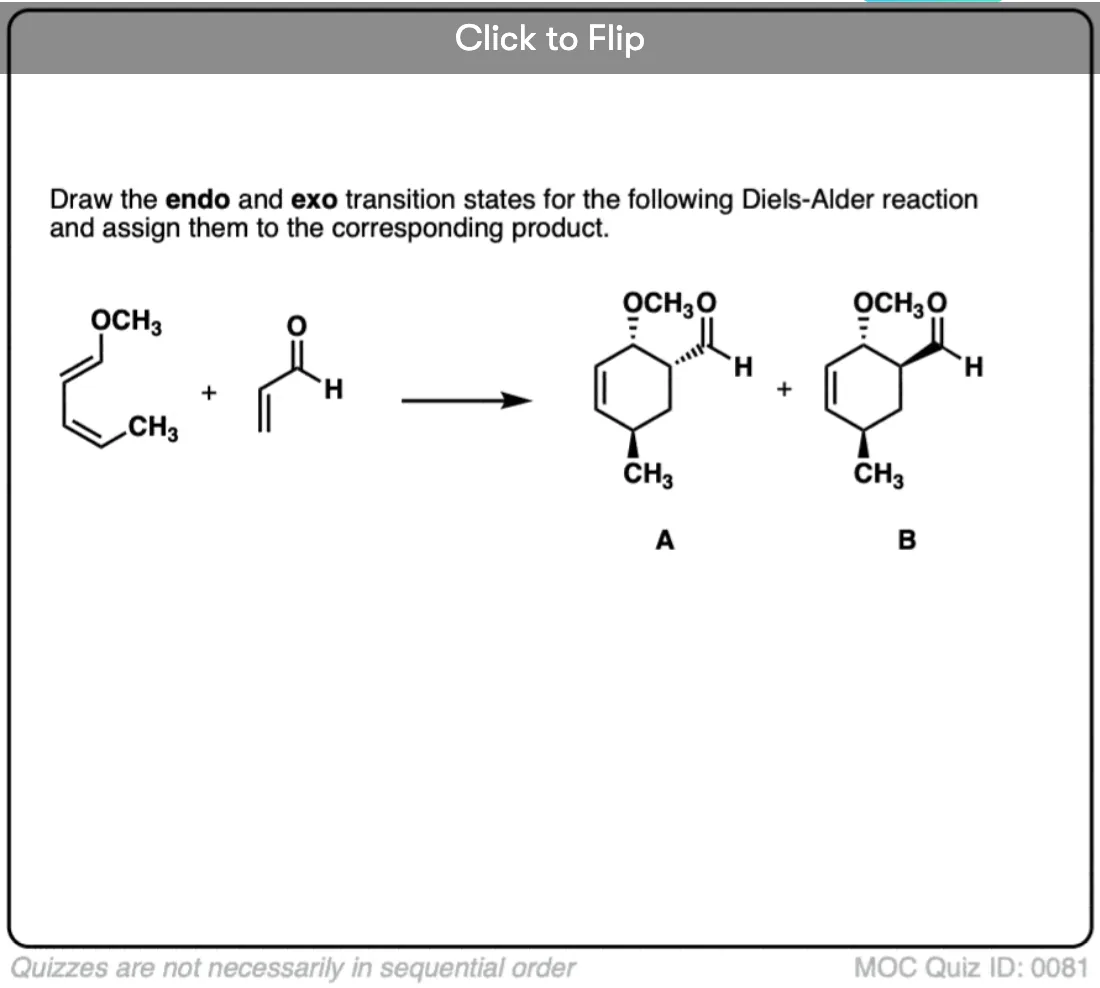
Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading
An important stereochemical feature of the Diels-Alder reaction is addressed by the Alder Endo Rule. This is based on empirical observations that if two isomeric products are possible, the one that has an unsaturated substituent(s) on the alkene oriented toward the newly formed cyclohexene double bond is the preferred product. The two alternative transition states are referred to as the endo and exo transition states.
- Untersuchungen über den Verlauf der Diensynthese
Kurt Alder, Gerhard Stein
Angew. Chem. 1937, 50 (28), 510-519
DOI: 10.1002/ange.19370502804
First paper by Alder describing what is now known as the Alder Endo Rule. After a “sandwich-like” preorientation of the reactants, the dienophile is added in such a way as to give a “maximum concentration” of double bonds in the transition state; according to Alder and Stein this includes not only the p systems directly involved in the reaction, but also those of the “activating ligands”. - ACCELERATION OF THE DIELS-ALDER REACTION BY ALUMINUM CHLORIDE
Peter Yates and Philip Eaton
Journal of the American Chemical Society 1960 82 (16), 4436-4437
DOI: 10.1021/ja01501a085 - Lewis acid catalysis of Diels-Alder reactions
K. N. Houk and R. W. Strozier
Journal of the American Chemical Society 1973, 95 (12), 4094-4096
DOI: 10.1021/ja00793a070 - Transition structures of the Lewis acid-catalyzed Diels-Alder reaction of butadiene with acrolein. The origins of selectivity
David M. Birney and K. N. Houk
Journal of the American Chemical Society 1990, 112 (11), 4127-4133
DOI: 10.1021/ja00167a005
Lewis acid complexation accentuates both the energy and orbital distortion effects of substitution on the dienophile, and enhanced both the reactivity and selectivity of the dienophile relative to the uncomplexed compound. The effects are well-modeled by computational studies on the transition-state structures. Prof. K. N. Houk (now at UCLA) is very well-regarded today for his work in computational chemistry. - Do Secondary Orbital Interactions Really Exist?
José I. García, José A. Mayoral, and Luis Salvatella
Accounts of Chemical Research 2000, 33 (10), 658-664
DOI: 10.1021/ar0000152 - The Source of the endo Rule in the Diels−Alder Reaction: Are Secondary Orbital Interactions Really Necessary?
José Ignacio García, José Antonio Mayoral, and Luis Salvatella
J. Org. Chem. 2005, 1, 85-90
DOI: 10.1002/ejoc.200400424
One of the proposed explanations for the Alder Endo Rule is secondary orbital interactions, and there is debate as to whether these actually exist or not. - Diels‐Alder reactions II: The Reaction Mechanism
Dr. J. Sauer
Angew. Chem. Int. Ed. 1967, 6 (1), 16-33
DOI: 10.1002/anie.196700161
Even though this review might be over 50 years old, it still contains very useful information, such as the influence of Lewis Acids on endo:exo selectivity. - Endo and Exo transition states in the Diels-Alder reaction
William C. Herndon and Lowell H. Hall
Tetrahedron Lett. 1967, 8 (32), 3095-3100
DOI: 10.1016/S0040-4039(00)90922-5
An very early theoretical study on Diels-Alder transition states, providing geometric arguments for the Alder Endo Rule. - Looking beyond the endo Rule in a Diels-Alder Discovery Lab
Ronald M. Jarret, Jamie New, Rebecca Hurley, and Laura Gillooly
Journal of Chemical Education 2001, 78 (9), 1262
DOI: 10.1021/ed078p1262
This paper from the Journal of Chemical Education shows how the Alder Endo Rule can be observed in a simple Diels-Alder reaction carried out by undergraduates. - The First General Enantioselective Catalytic Diels−Alder Reaction with Simple α,β-Unsaturated Ketones
Alan B. Northrup and and David W. C. MacMillan
Journal of the American Chemical Society 2002 124 (11), 2458-2460
DOI: 10.1021/ja017641u
In this paper, D. W. C. MacMillan’s research group investigated cycloadditions between ethyl vinyl ketone, EtCOCH=CH2, and several simple dienes. As a rule, endo selectivity was not very high. They reasoned that making the dienophile more electron-poor should improve selectivity (and reaction rate, and yield) and this might be achieved by converting the carbonyl group, C=O, into an iminium ion, C=NR2+. They also reasoned that it should be possible to produce the iminium ion catalytically by combining the ketone with a small amount of acid and a small amount of a chiral amine. This area is now known as organocatalysis, and a tremendous amount of work now being done in this area. - The Diels Alder Reaction
The late Hans Reich (U. Wisconsin-Madison) has a website full of useful information on organic chemistry, including this page on the Diels-Alder reaction.
superb!!! thank you for such helpful explanation
Very good explained and is really helpful…i must appriciateyou for publishing such beneficial addition to organic chemistry
Really it’s helpful…thank you for publishing such beneficial topics on organic chemistry
OK, thank you Pompi. Always looking for suggestions to make it better.
Absolutely brilliant!!
Thanks for putting in so much effort and helping us out.
Thanks Gaurav. Glad you find it helpful.
This is very helpful information. Not explained well by my lecturer. Thank you.
Glad this was useful Jordan!