Reactions of Aromatic Molecules
Nucleophilic Aromatic Substitution (2) – The Benzyne Mechanism
Last updated: May 28th, 2026 |
In this article we’ll discuss nucleophilic aromatic substitution, but with a new twist; a nucleophilic aromatic substitution that passes through a strange-looking intermediate called an aryne (a generic term for a family of molecules that includes benzyne).
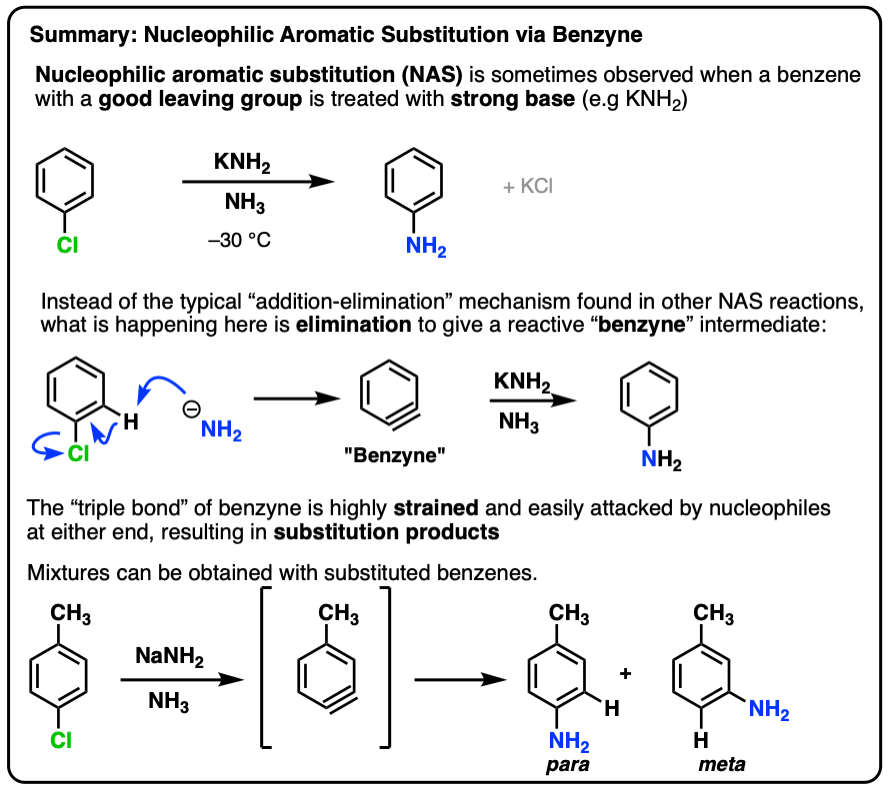
Table of Contents
- Nucleophilic Aromatic Substitution – A Quick Recap
- A “Nucleophilic Aromatic Substitution” In Name, But By A Different Mechanism
- The “Benzyne” Intermediate
- Reaction of Substituted Benzyne – “Arynes”
- Benzyne Undergoes Diels-Alder Reactions
- The Structure of Benzyne
- Summary: Nucleophilic Aromatic Substitution via Benzyne
- Notes
- Quiz Yourself!
- (Advanced) References and Further Reading
1. Quick Recap: Nucleophilic Aromatic Substitution
Previously [see: Nucleophilic Aromatic Substitution] we saw that electron-poor aromatic rings containing a leaving group can undergo substitution with electron-rich nucleophiles. We saw that the mechanism proceeds through addition of a nucleophile to the aromatic ring (via an electron-rich intermediate) followed by loss of a leaving group, in a process sometimes called, “addition-elimination”.
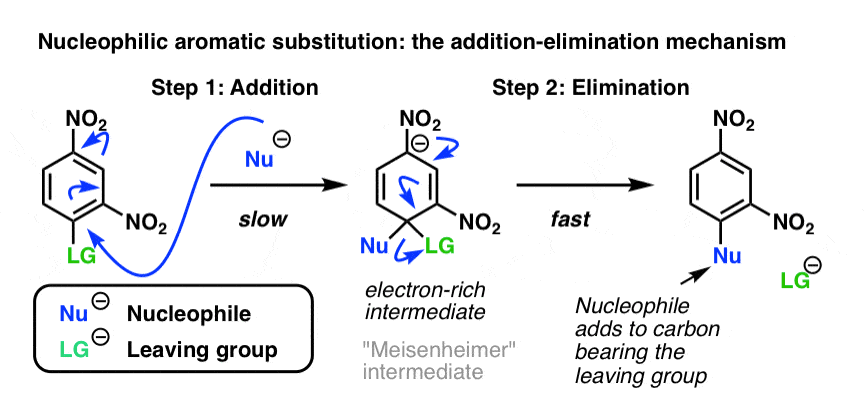
Importantly, the only substitution product is the one where the nucleophile ends up attached to the same carbon as that bearing the leaving group. (This differentiates it from electrophilic aromatic substitution, where a mixture of ortho-, para– and meta- products can be obtained.)
2. A “Nucleophilic Aromatic Substitution” In Name, But By A Different Mechanism
Although the “addition-elimination” mechanism for nucleophilic aromatic substitution has been known since at least 1902 , it became increasingly clear in the first half of the twentieth century that certain reactions classified as “nucleophilic aromatic substitution” appeared to proceed through a different mechanism altogether.
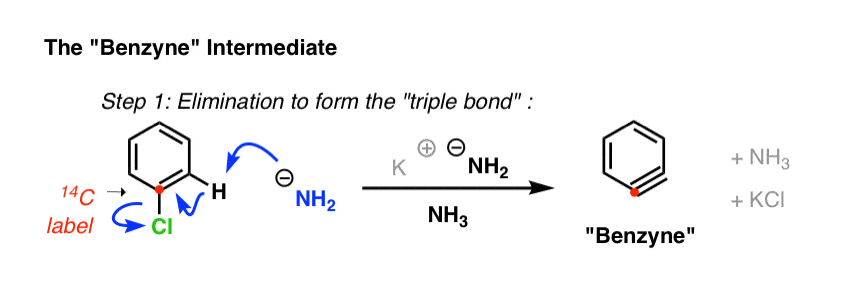
For example, it was found that treating chlorobenzene with sodium amide (NaNH2) in liquid ammonia (boiling point = –33°C) resulted in the rapid formation of aminobenzene (“aniline”):
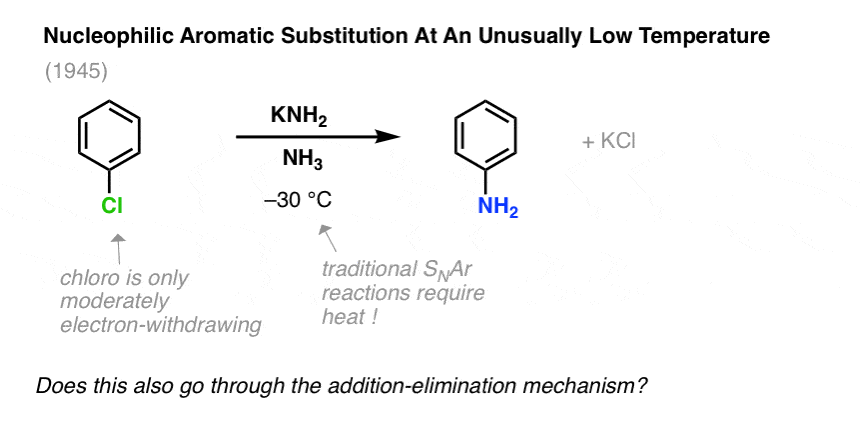
An addition-elimination mechanism here doesn’t seem right, considering that nucleophilic aromatic substitution reactions with far stronger electron withdrawing groups (e.g. NO2, rather than Cl) require higher temperatures and longer reaction times.
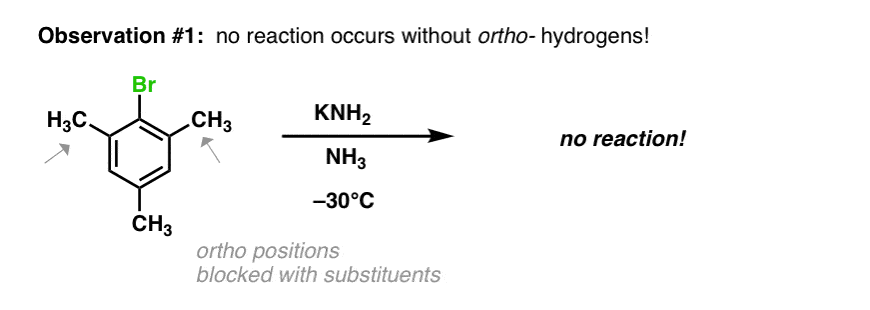
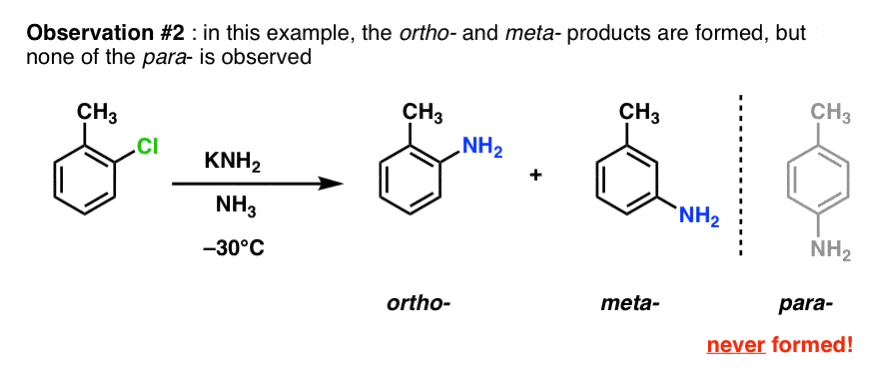
Another observation was that no reaction occurred under these conditions when the ortho- positions were attached to alkyl groups. A hydrogen is necessary at one of these positions for the reaction to proceed.
(note – NaNH2 and KNH2 can be considered to be essentially the same for our purposes)
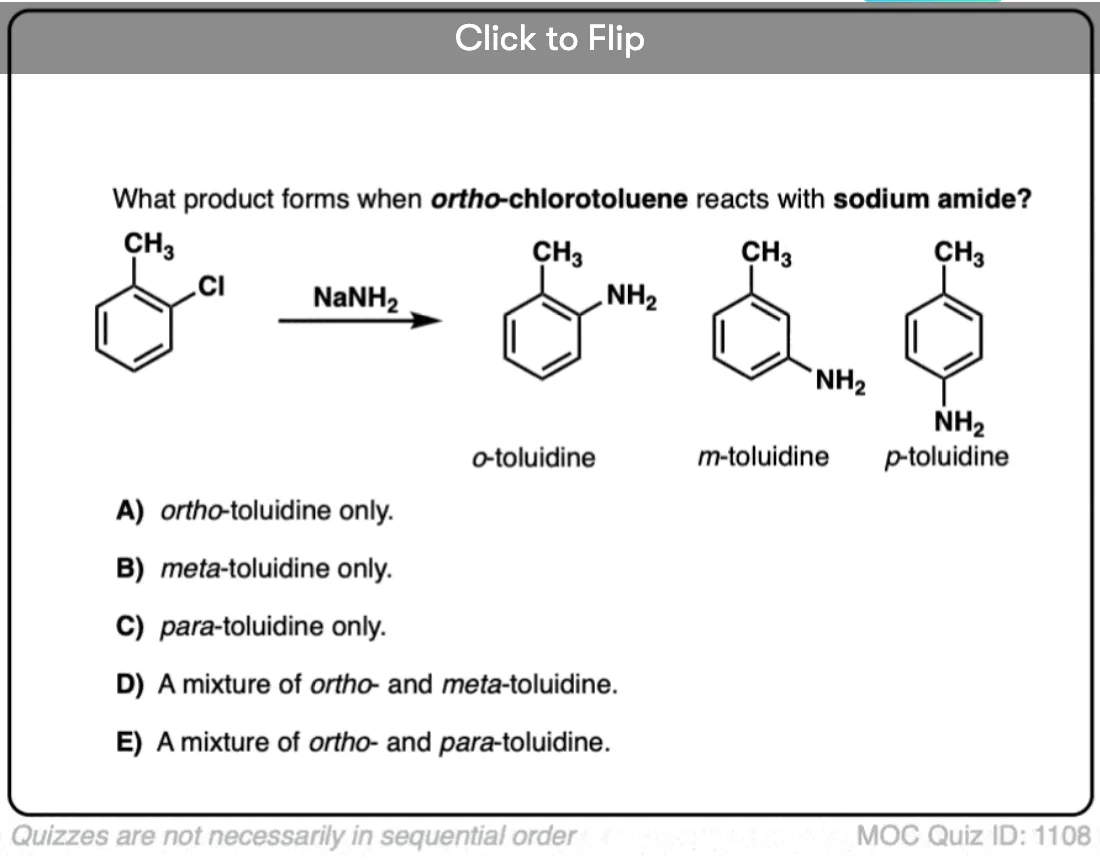
A second observation was that in the case below only the ortho- and meta- products formed, and never the para– .
3. The Benzyne Intermediate
Various intermediates were proposed to explain these results, but then in 1953 John D. Roberts (then at MIT) nailed it by publishing one of the most elegant chemical experiments of all time.
He and his team synthesized chlorobenzene but with a special difference: the carbon attached to the chlorine was a radioactive isotope of carbon (14C), not carbon (12C).
This radioactive carbon atom served as an atomic “label”, which allowed them to conclusively determine if substitution happened exclusively at the carbon bearing the leaving group. (how? [Note 4])
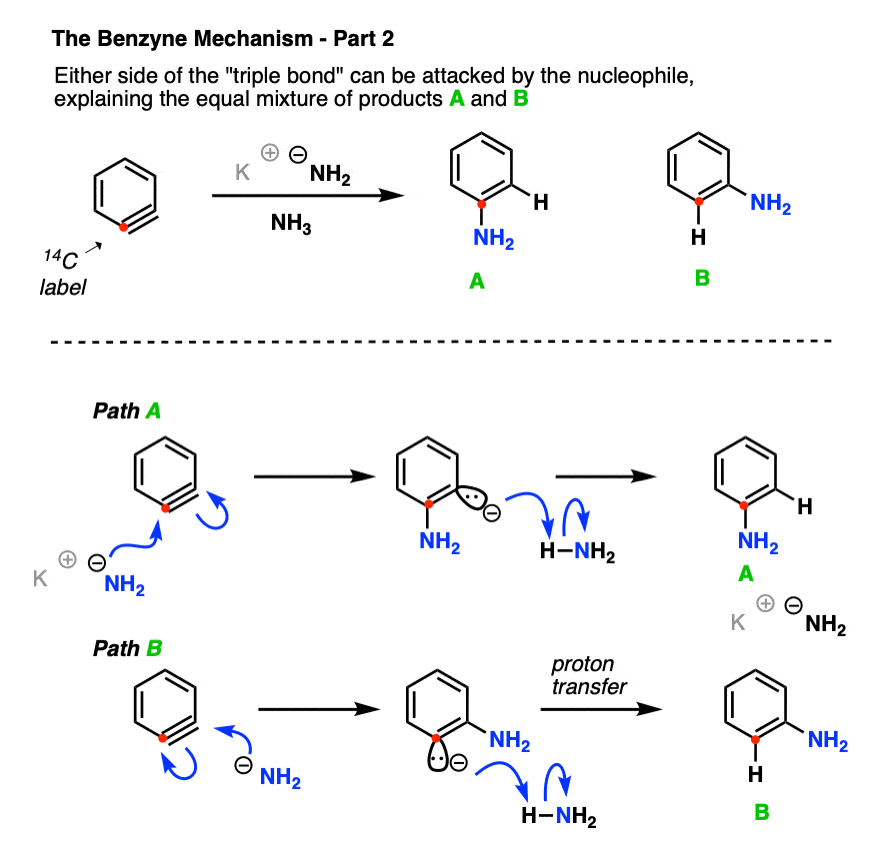
Roberts’ group carried out the reaction under conditions reported previously, and found that about 50% of the product ended up with the NH2 attached to the labelled carbon, and the other 50% had the NH2 on the carbon adjacent to the label.
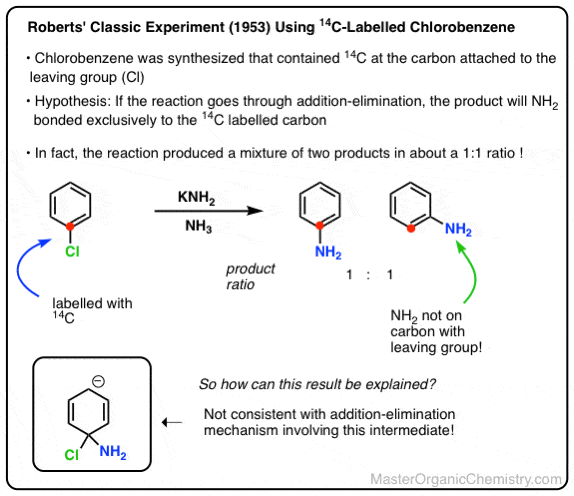
This is not consistent with an addition-elimination mechanism!
In fact, the roughly 50:50 ratio of products implies the involvement of a symmetrical intermediate which is attacked equally on either side.
Roberts’ proposal – which has stood the test of time – was the involvement of a short-lived intermediate bearing a carbon-carbon triple bond: “Benzyne” !
At first glance, this seems crazy. A triple bond in an aromatic ring?
Well, it’s not quite a true triple bond in the way that we’re familiar with (i.e. with alkynes). Instead of an overlap between two 2p orbitals (as in an alkyne) the “triple bond” is formed through overlap of two adjacent sp2 orbitals in the plane of the ring (i.e. at right angles to, and completely independently of, the aromatic pi system).
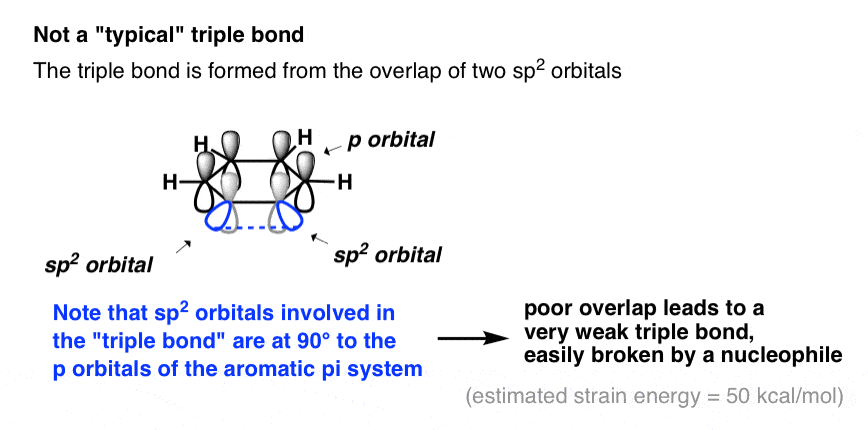
Since these orbitals actually point away from each other, the overlap between them is poor, resulting in a “triple bond” that is actually very weak.
The strain energy of benzyne has been estimated to be about 50 kcal/mol – more strained than cyclopropane (28 kcal/mol), and only slightly less strained than cyclopropene (54 kcal/mol).
An intuitive way to think about it is to imagine the involvement of two resonance structures (far left and far right, below) that make strong (and equal) contributions to the overall resonance hybrid, such that both carbons can be considered “electrophilic”.
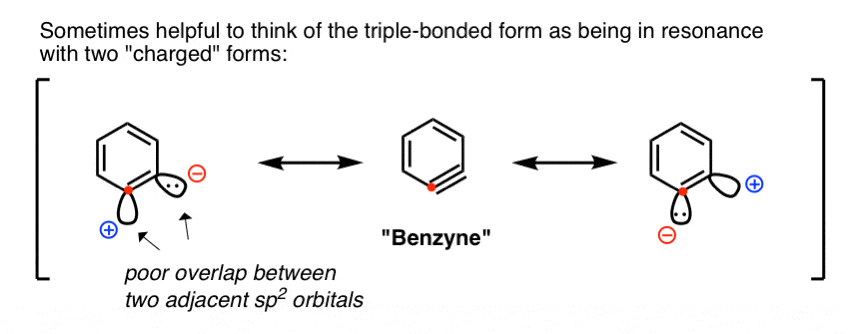
[A more rigorous way to treat it is from a molecular orbital perspective – a weak bond results in a low-energy LUMO, and therefore a lower energetic barrier to attack by nucleophiles].
However strange it might look, the benzyne intermediate explains all of these important observations, and more.
- In the first step (elimination) a strong base removes a hydrogen from the carbon adjacent to that bearing the leaving group, resulting in an elimination reaction that forms the triple bond. This explains why no reaction occurs when both positions adjacent to the leaving group lack hydrogen!
- In the second step (addition), attack of the can come at either side of the triple bond, resulting in about a 1:1 mixture of the product with NH2 attached to the labelled carbon (A) and NH2 adjacent to the labelled carbon (B).
Although it’s tempting to use –NH2 as nucleophile, the more likely nucleophile here is the solvent, NH3, which readily reacts with the extremely reactive benzyne intermediate. After attack of NH3, proton transfer occurs to result in the neutral product. [note that although intramolecular proton transfer is shown below, it’s also possible to show it occurring through an intermolecular mechanism]
4. Reactions Of Substituted Benzyne (“Arynes”)
What happens when a substituent is present on benzyne? [Note: Just like substituted compounds of benzene are called “arenes”, substituted benzynes are called “arynes”. ]
When a substituent is present, an unsymmetrical aryne will result, and at least two products (and sometimes three) can potentially form. In the example below, attack of NH3 at carbon A results in the ortho product, and attack of NH3 at carbon B results in the meta product.
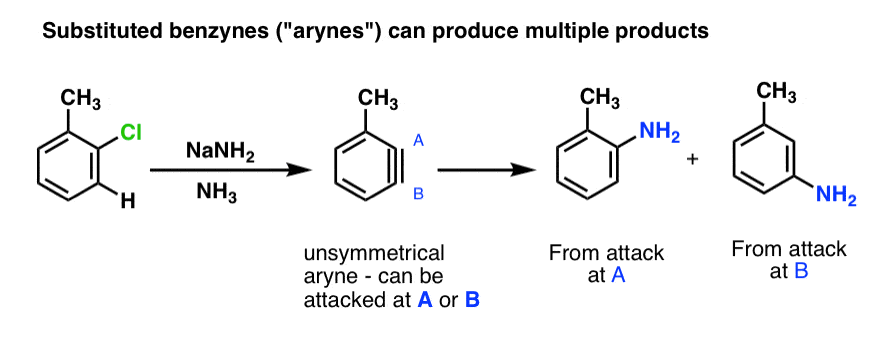
So how do substituents on the ring affect addition to the triple bond?
Since the aromatic pi-system is at right angles to the triple bond, what’s NOT relevant is the ability to donate a lone pair to the ring (like OCH3) , or accept a lone pair from it (like NO2).
However, inductive effects are still relevant (i.e. electron-withdrawing effects that occur through single bonds).
Why? Addition to the triple bond creates a negative charge on carbon, and electron-withdrawing groups stabilize negative charge: the closer, the better.
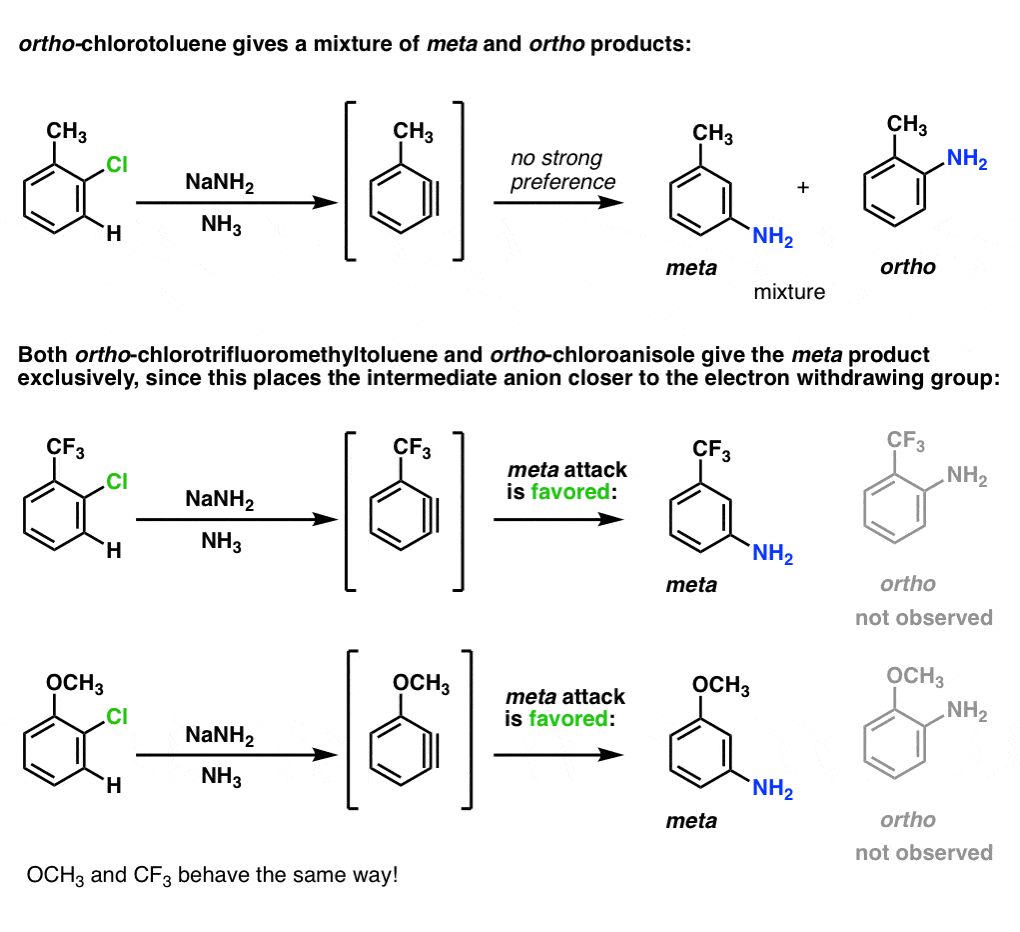
So a key principle in the addition of nucleophiles to arynes is that addition tends to happen so as to place the negative charge closer to an electron-withdrawing substituent.
Two key examples.
- When the triple bond is between the meta and para carbons, attack will favor the para product, since the negative charge ends up on the meta carbon (closer than para).
- When the triple bond is between the ortho and meta carbons, attack will favor the meta product, since the negative charge ends up on the ortho carbon (closer than meta)
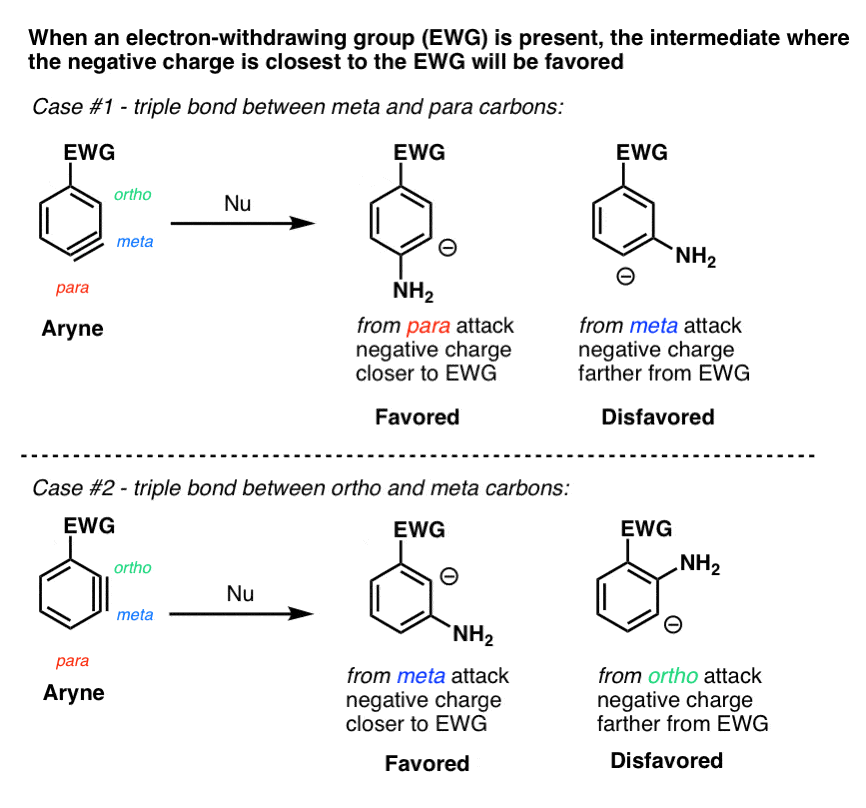
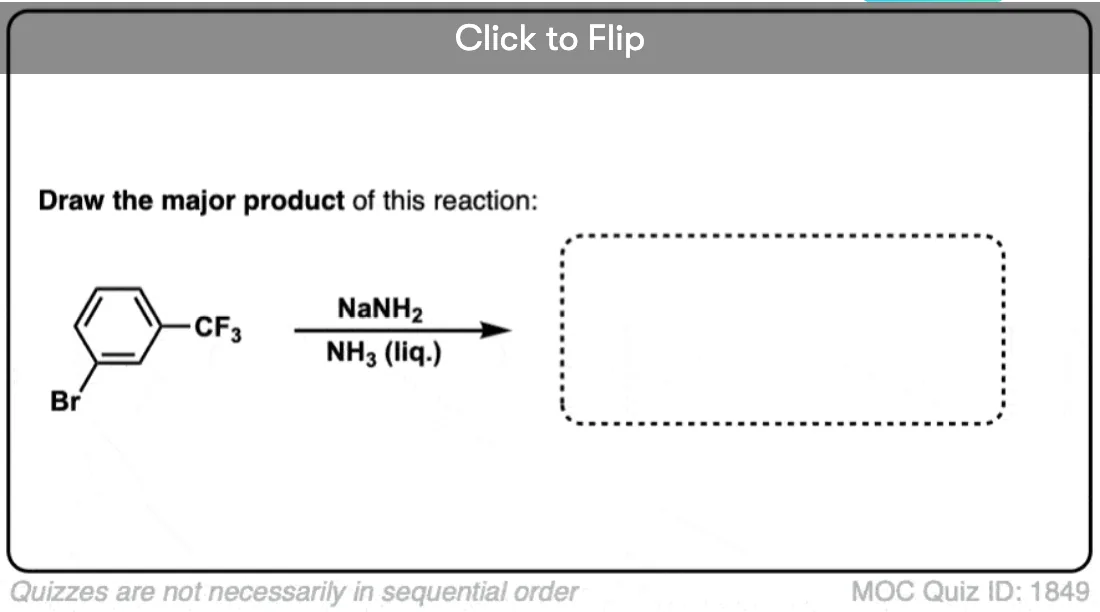
Note that this only applies for electron-withdrawing groups like CF3, not electron-donating groups like CH3 or alkyl. What’s interesting is that OCH3 behaves like an electron-withdrawing group in these examples, since the oxygen lone pairs can’t interact with the triple bond.
See the [Note 1] for some more examples.
5. Benzyne Undergoes Diels Alder Reactions
Benzyne can also react with dienes in Diels-Alder reactions. The triple bond of benzyne acts as a dienophile:
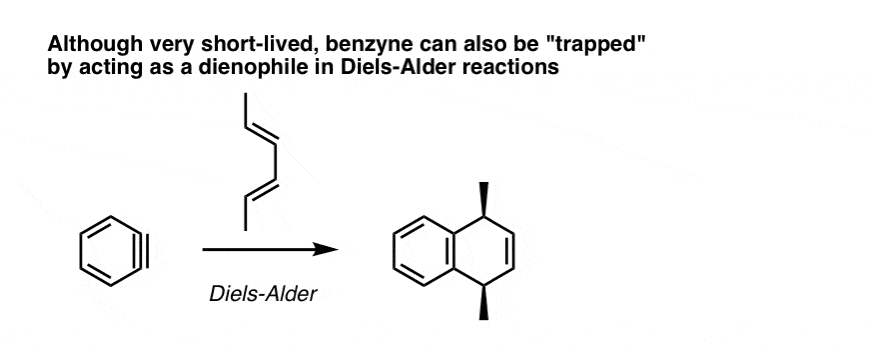
6. The Structure Of Benzyne
For many years benzyne defied attempts at isolation and characterization, its existence inferred through trapping experiments with nucleophiles or dienes. It lives fast and dies young.
Eventually, benzyne was trapped at 6K (i.e. six degrees above absolute zero) in frozen argon and studied through various spectroscopic methods. The triple bond is about 1.26 Å, longer (and weaker) than that in ethyne (1.20 Å) and the adjacent C-C bond is about 1.38 Å , slightly shorter than that in benzene.
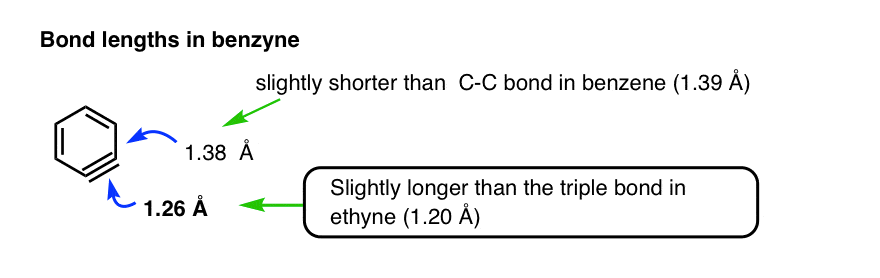
(See reference). All the other C-C bonds are within 0.01 Å of the bond lengths in benzene.
7. Summary: Nucleophilic Substitution Reactions Via Benzyne
There is one final type of aromatic substitution reaction for us to consider – a family of reactions called the Sandmeyer reaction that uses nitrogen gas (N2) as the leaving group. More on that next time.
Next Post: Reactions of Diazonium Salts and The Sandmeyer Reaction
Notes
- Some other examples of substitution
- Methods for generating benzyne
- Roberts’ degradation study
Some other examples of substituted benzynes
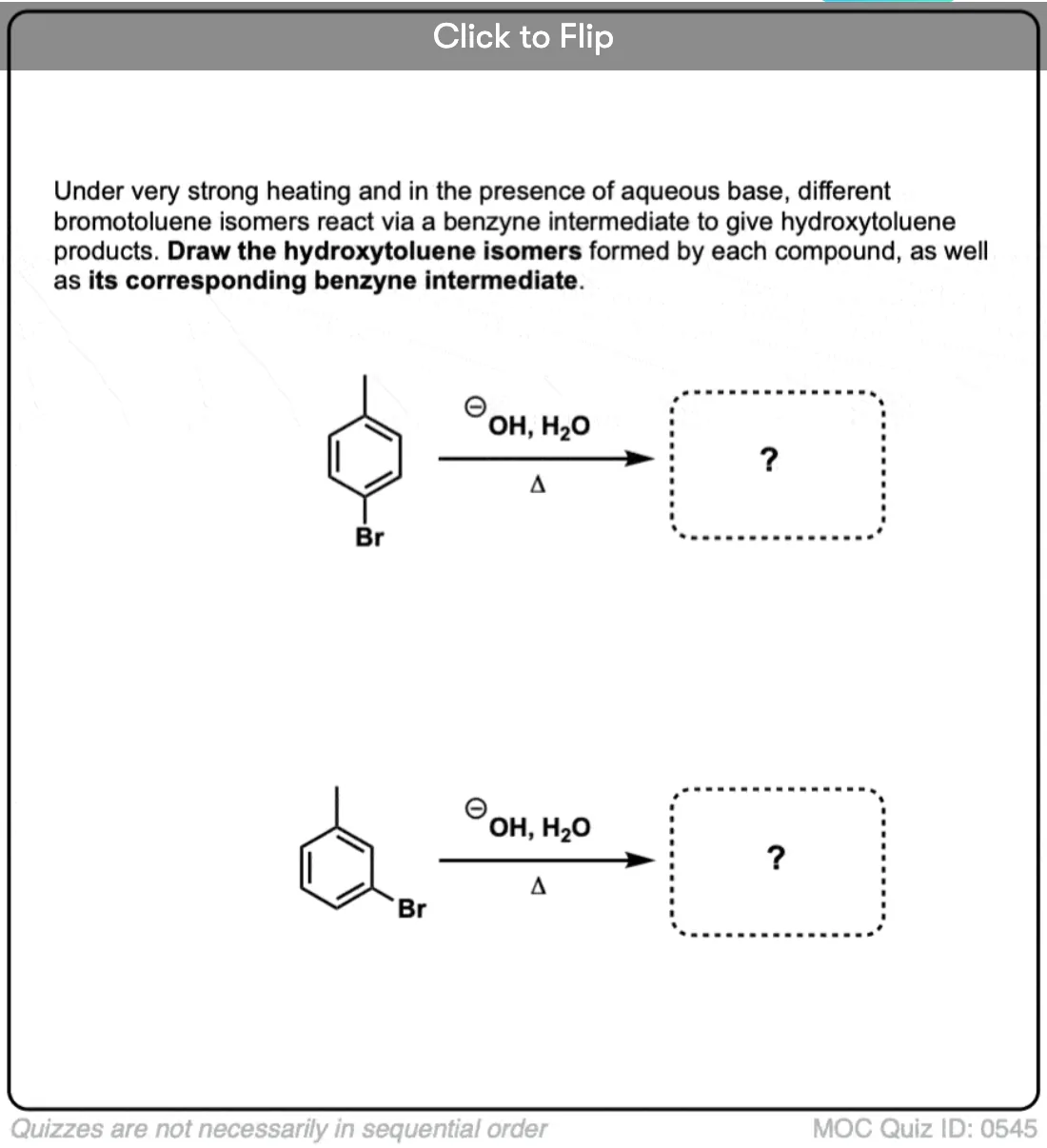
When an aryne is made from p-chlorotoluene, a mixture of para and meta products is obtained.
When an electron-withdrawing substituent like CF3 is used instead of CH3, more of the para is formed – about 60:40, favoring the para. (Statistics would favor the meta by 2:1).
This is because attack at the para position places the negative charge closer to the electron withdrawing group.
Interestingly, OCH3 gives about the same ratio as CF3 (60:40). This means that oxygen is acting purely as an electron-withdrawing group, not a pi-donor. (Further proof that the oxygen lone pair can’t interact with the triple bond. )
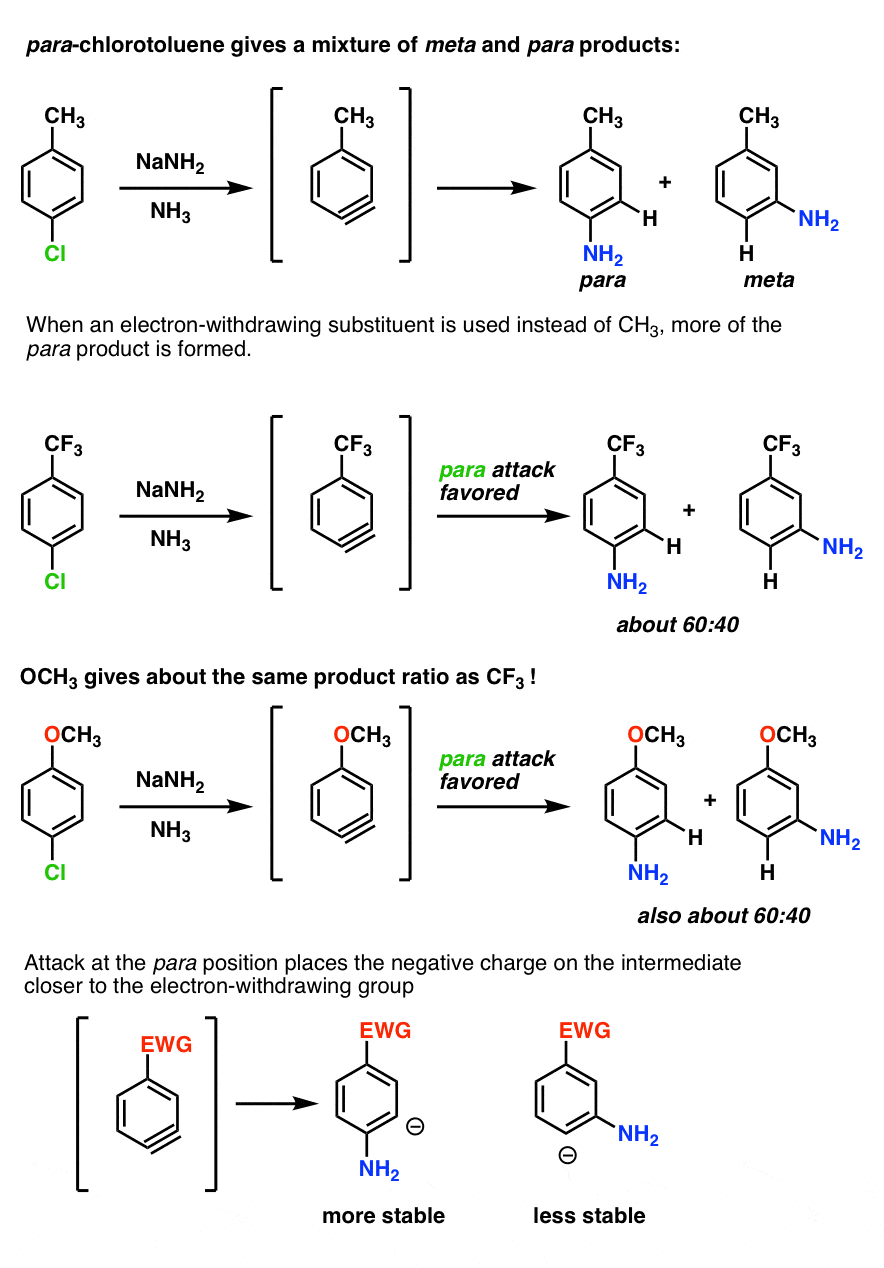
With the ortho-series, the results are even more dramatic. With CH3, about an equal mixture of ortho and meta products is obtained. With CF3 and OCH3, only the meta is observed.
Some ways of generating benzyne
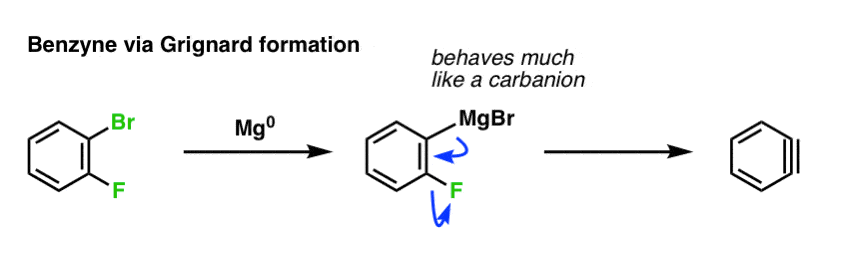
Benzyne can be generated in several other ways besides deprotonation with a strong base like NaNH2 or KNH2.
Grignard reagents behave much like carbanions. So a different path to benzyne formation is to start with a dihalobenzene like o-bromofluorobenzene and treat it with magnesium metal. The Grignard forms, and then this “anion” can displace even a good leaving group like fluorine to give the triple bond:
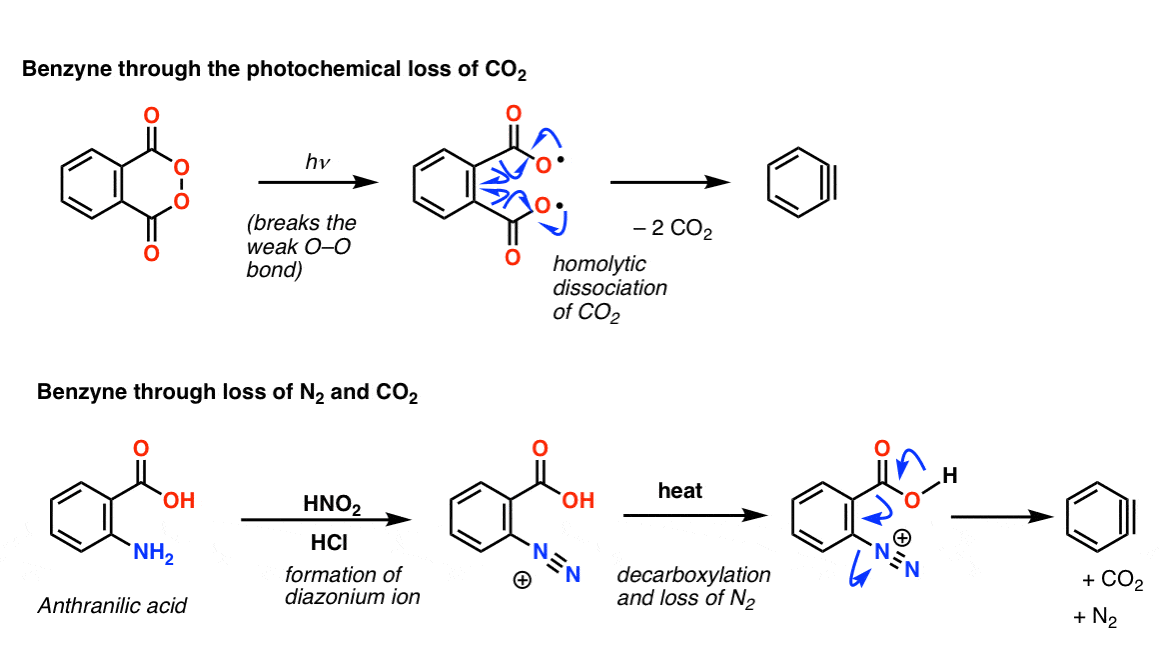
Another path is to generate a leaving group that can leave irreversibly as a gas, like CO2 or N2.
In the first reaction below, UV light breaks the weak O-O bond, and then homolytic dissociation results in loss of two CO2 molecules to give benzyne.
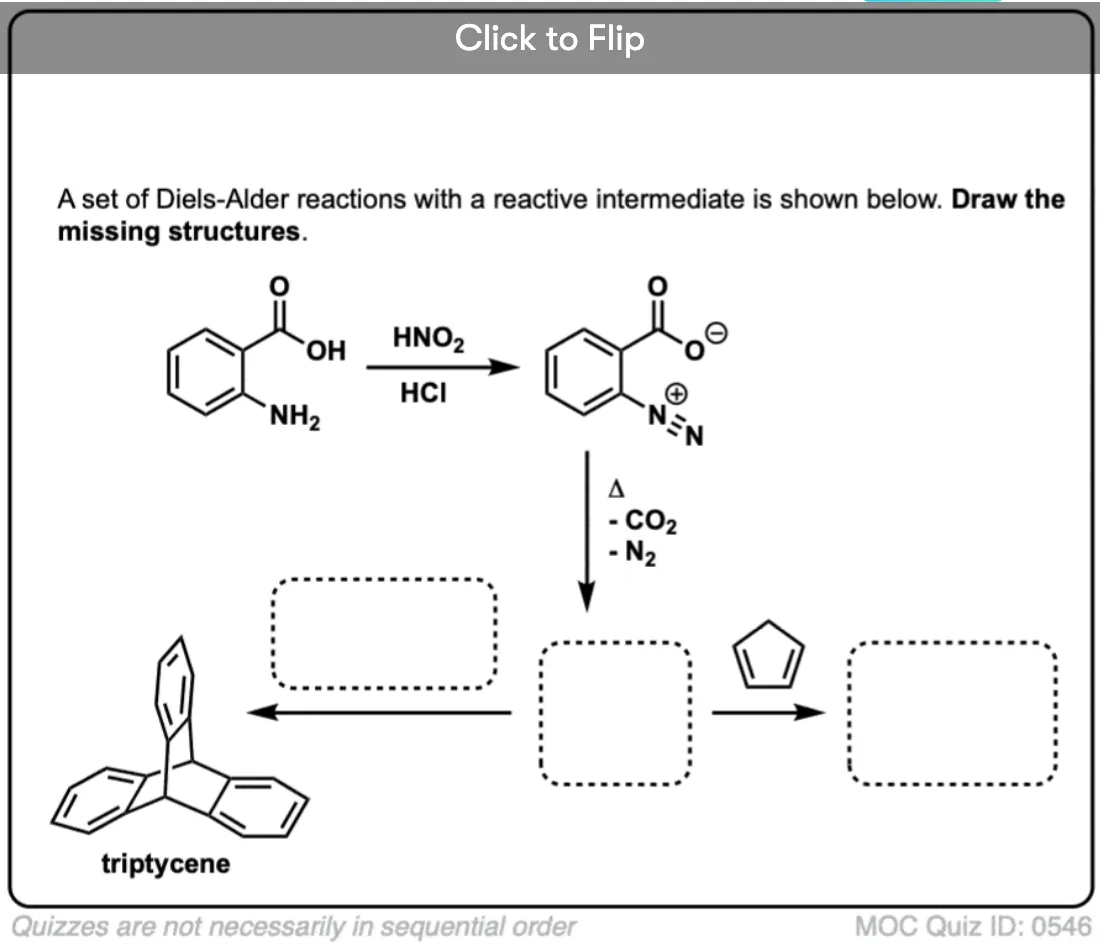
In the second example, commercially available (and cheap) anthranilic acid is converted into a diazonium ion with HNO2 and HCl. Heat results in loss of CO2 and N2:
This is just the tip of the iceberg. For more reading, this presentation on arynes by Eric Welin of the Macmilllan group at Princeton is an excellent introduction.
Degradation study.
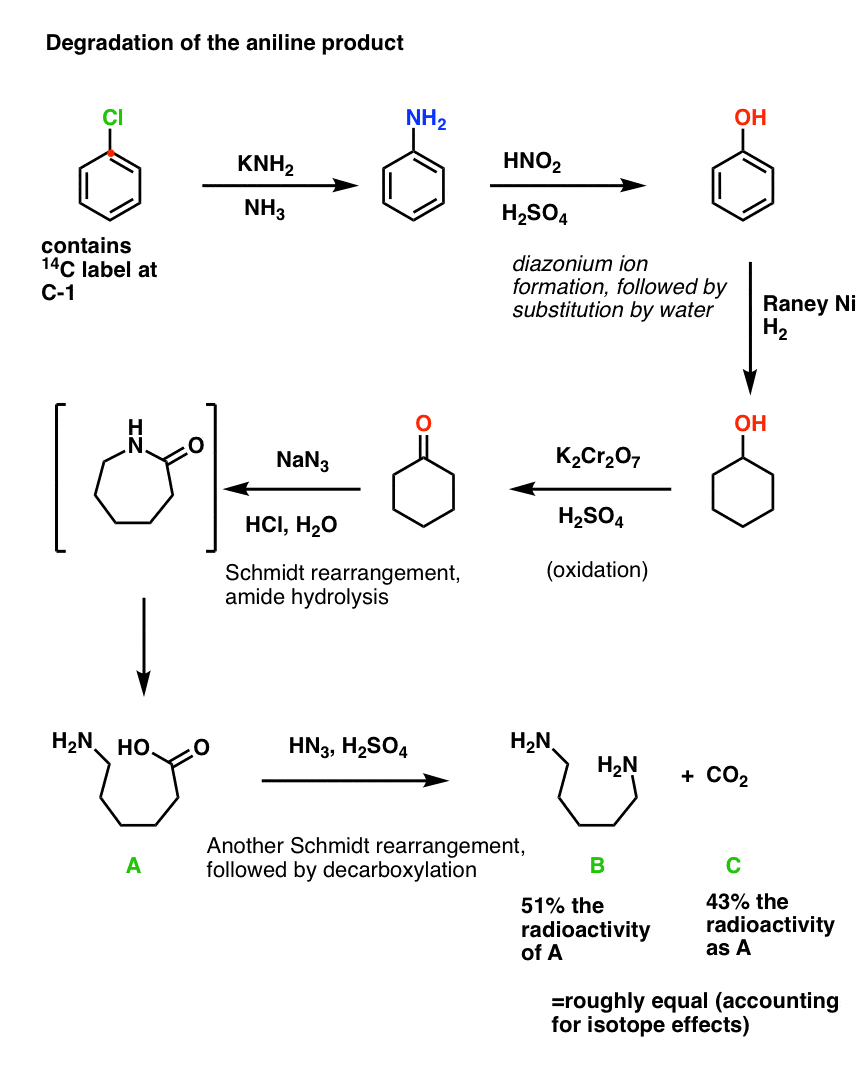
Today, the results of Roberts’ labelling experiment could be determined in about 5 minutes through a technique called Nuclear Magnetic Resonance, or NMR.
But in 1954, this technique didn’t exist. So it was necessary to determine the ratio by breaking the products down into simpler components via well-understood chemical reactions, a technique known as “degradation”.
Aniline was converted into phenol through diazonium salt formation and substitution with water. Hydrogenation with Raney nickel under high pressure gave cyclohexanol, which was then oxidized to cyclohexanone with chromic acid. A Schmidt reaction converted cyclohexanone into a lactam, which was hydrolyzed to the free acid. A second Schmidt reaction then liberated CO2, which was trapped with barium hydroxide. The radioactivity of the CO2 and the diamine products were found to be roughly equal (and each about half of the total reactivity of the acid precursor), thus confirming that attack of the nitrogen occurred at two different carbons.
Our chemical ancestors had to perform the labor of Job just to get a structure.
The person who did more than anyone else to develop and popularize the use of NMR as a technique for structure determination of organic compounds, and thereby free us from this misery?
John Roberts, who began working on it shortly after this benzyne study was published.
Quiz Yourself!

Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
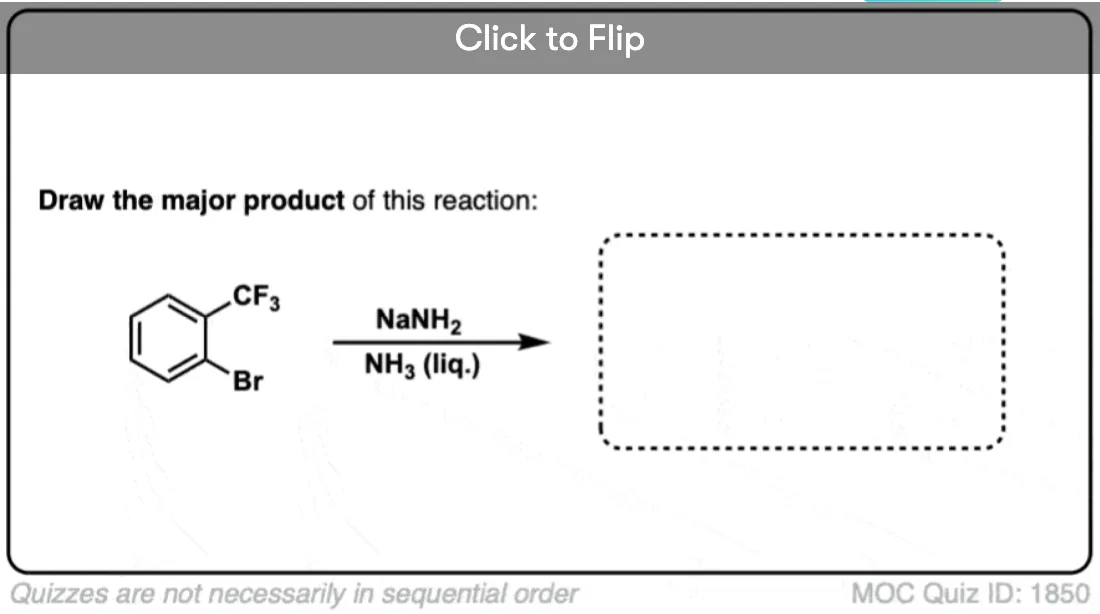
Become a MOC member to see the clickable quiz with answers on the back.
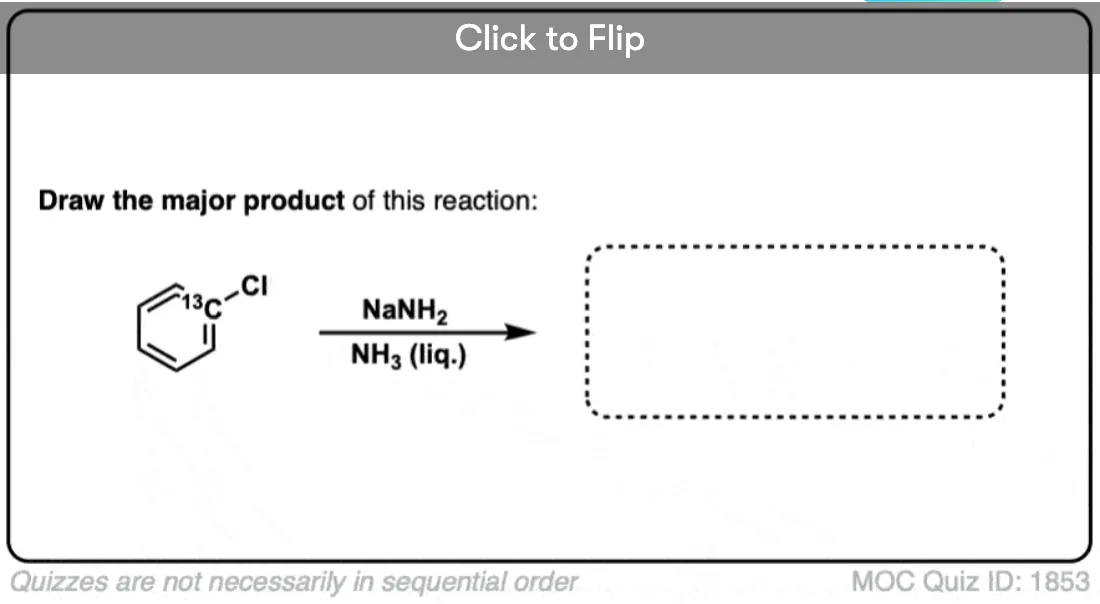
Become a MOC member to see the clickable quiz with answers on the back.
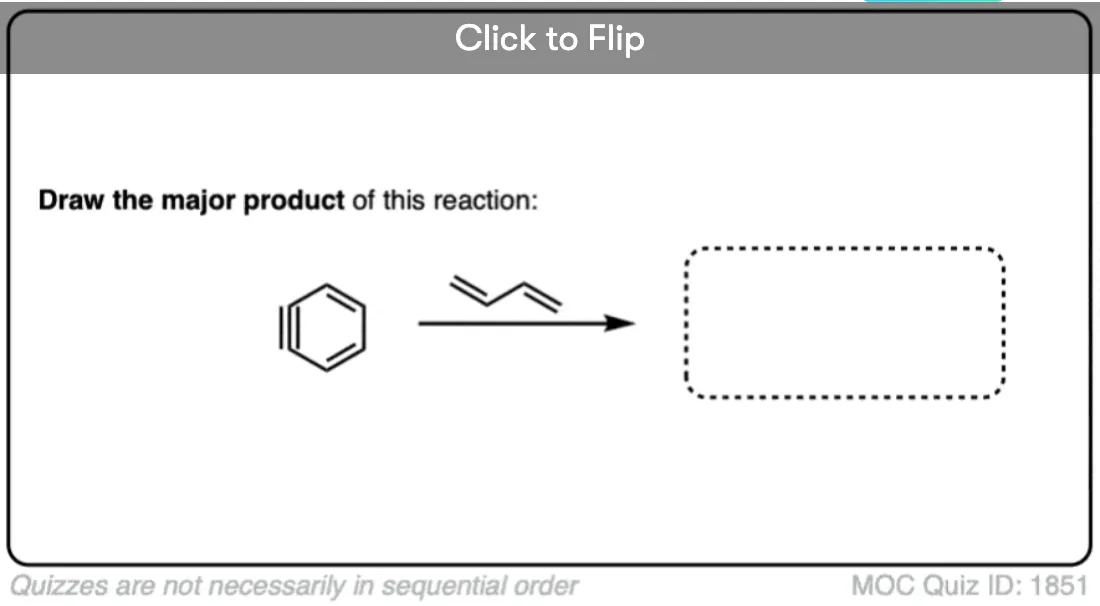
Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References And Further Reading
In these first two papers, Nobel Laureate Georg Wittig describes reactions of o-fluorophenyllithium and proposes they go through a zwitterionic intermediate (although in paper 2 he does also use a triply bonded structure for o-benzyne).
- Über die Bildung von Diphenyl aus Fluorbenzol und Phenyl‐lithium (IV. Mitteil. über Austauschreaktionen mit Phenyl‐lithium)
Georg Wittig, Gustav Pieper, Gerhard Fuhrmann
Ber. 1940, 73 (11), 1193-1197
DOI: 10.1002/cber.19400731113 - Phenyl-lithium, der Schlüssel zu einer neuen Chemie metallorganischer Verbindungen
Georg Wittig
Naturwissenschaften November 1942, Volume 30, Issue 46–47, pp 696–703
DOI: 1007/BF01489519These papers are classics by Prof. J. D. Roberts in which he used 14C-labeled chlorobenzene and KNH2 to prove the intermediacy of benzyne due to scrambling of the 14C label after the reaction. - REARRANGEMENT IN THE REACTION OF CHLOROBENZENE-1-C14 WITH POTASSIUM AMIDE
John D. Roberts, Howard E. Simmons Jr., L. A. Carlsmith, and C. Wheaton Vaughan
Journal of the American Chemical Society 1953 75 (13), 3290-3291
DOI: 1021/ja01109a523 - The Mechanism of Aminations of Halobenzenes
John D. Roberts, Dorothy A. Semenow, Howard E. Simmons Jr., and L. A. Carlsmith
Journal of the American Chemical Society 1956 78 (3), 601-611
DOI: 1021/ja01584a024 - TRIPTYCENE
Georg Wittig
Org. Syn. 1959, 39, 75
DOI: 10.15227/orgsyn.039.0075
This procedure in Organic Syntheses, a source of reliable and independently tested experimental synthetic organic chemistry procedures, is also from Georg Wittig and is a classical method for preparing benzyne. - FLUORIDE-INDUCED 1,2-ELIMINATION OF O-TRIMETHYLSILYLPHENYL TRIFLATE TO BENZYNE UNDER MILD CONDITIONS
Yoshio Himeshima, Takaaki Sonoda, Hiroshi Kobayashi
Lett. 1983, 1211-1214
DOI: 10.1246/cl.1983.1211
This is an extremely influential paper in organic chemistry, since this chemistry enables the generation of benzyne under extremely mild conditions (room temperature or below). - THE DIRECT ACYL-ALKYLATION OF ARYNES [PREPARATION OF METHYL 2-(2-ACETYLPHENYL)ACETATE]
David C. Ebner, Uttam K. Tambar, and Brian M. Stoltz
Org. Syn. 2009, 86, 161
DOI: 10.15227/orgsyn.086.0161
An example of a modern synthetic procedure involving benzyne, using the chemistry developed by Kobayashi (ref. 6). Prof. Brian Stoltz (Caltech) has developed (and continues to develop) many interesting synthetic methodologies using benzyne. - Indolyne Experimental and Computational Studies: Synthetic Applications and Origins of Selectivities of Nucleophilic Additions
G-Yoon J. Im, Sarah M. Bronner, Adam E. Goetz, Robert S. Paton, Paul H.-Y. Cheong, K. N. Houk, and Neil K. Garg
Journal of the American Chemical Society 2010 132 (50), 17933-17944
DOI: 10.1021/ja1086485
Prof. Neil Garg (UCLA) did his PhD with Prof. Stoltz and has extended the benzyne concept to other heterocycles, such as indole, notably developing chemistry based on ‘indolyne’. - One Century of Aryne Chemistry
Hans Henning Wenk Dr., Michael Winkler Dipl.‐Chem., Wolfram Sander Prof. Dr.
Chem. Int. Ed. 2003, 42 (5), 502-528
DOI: 10.1002/anie.200390151
Nice review on aryne chemistry.
‘,’Nucleophilic Aromatic Substitution (2) – The Benzyne Mechanism
I have a little problem with the fact that electron delocalization via resonance is irrelevant . I feel like it should have a significant impact . Even though the negative charge on the carbon after nucleophilic attack cant be stabilized through conjugation , the electron density at ortha and para positions for electron withdrawing groups should be less than that without them. This reasoning stems from the electrophilic substitution reactions. So why doesnt this have any effect
In that text of benzyne mechanism given that nucleophilic attack on o chloro toulene forms meta and ortho products both without favoring anyone of them.
Pls confirm what should be major as in my exam ortho was given pls clarify!
There is no strong preference. A mixture is obtained. If your instructor says ortho is favored exclusively over meta I would love to hear why, because that’s not what’s observed.
This is a very informative post! However there’s a doubt with CF3 substituent. Cf3 shows negative hyperconjugation which doesn’t act at meta positions(only at ortho and para)..So shouldn’t that stabilize the anion/negative charge at para position more as opposed to at meta position. Shouldn’t -Nh2 then be attached to meta position in the major product and not to para position then..?
How protic and aprotic solvent effect benzyne formation. (Brief explanation needed)
So wellexplained and helpful! Your clear and lucid language really helps me a lot in understanding many important topics. Whenever I look for doubt clearance with respect to organic chemistry and I find your post, I know they’re gonna be well looked after. With tons of thanks, from India.🙏🙏
Thank you
Thankyou so much. All content in this website is very informative
Thank you sir.It helps me to understand this concept which is very important for indian competitive exams.
Excellent ..the way you divided & explained the concept🤩
Well done for the teaching☺☺
Informative post.. just a quick question… how do we decide if a reaction proceeds in the normal Addition Elimination Mechanism or the Benzyne Mechanism.. any parameters like temperature/reagent, etc?
Look for the presence of strong base (NaNH2) in the case of aryne formation.
Amazing content! Thank you!
I had some confusion however ; in the question posed in the flashcard (“Quiz Yourself!”) the second case deals with a substituent at meta position. The solution gives two possible Benzyne intermediates ; one with the triple bond between m,p and one at o,p. How do we decide/expect which one will be major?
Thanks a ton .Amazing as always. Probably the best few pages on benzyne. Salute to you sir
Nice post!! But I have a doubt…
Why doesn’t resonance occur in benzyne? Is it because it loses aromaticity?
Is that the reason why we don’t consider resonance effect of the substituent while deciding the product?
The “triple bond” in benzyne is in orbitals that are at 90° to the aromatic pi system. They are not able to interact with the orbitals that comprise the aromatic ring.
love From INDIA..
in our culture teachers are god to us..
u proved to be one definitely!
I don’t understand why all my textbooks (Clayden, Yurkanis, Vollhardt) show that the NH2- is the nucleophile and NEVER the NH3! It makes so much sense with the NH3 and your explanation. Even my professor taught it with the NH2-. Now I don’t know how I will write it in my test. Do you have any book that talks about this?
Thanks for such a great website by the way! Greeting from Germany
Just to be sure, the rate determining step is the second one, with the attack of the nucleophile on the aryne intermediate, right? I couldn’t find a mention of that in the post.
No, rate determining step is formation of the aryne (deprotonation). The attack is fast.
excellent I always use these helpful posts and recommend it to my friends, all the posts are great, thank you very much
Excellent post, as expected. One minor thing, though:
“Benzyne Undergoes Diels Alder Reactions
dienes in a Diels-Alder reaction. The triple bond of benzyne acts as a dienophile:”
It looks like the 1st part of the 1st phrase is missing.
Shoot. Thank you! Fixed.
Great post, James!
I just wanted to point out what is probably a copy/paste error in your discussion of the regioselectivity of substituted arynes. In case #2 (when the triple bond is between the ortho and meta carbons), you’ve drawn both products as having the same structure, and the accompanying text in the ChemDraw is incorrect.
Also, you might be interested in this nice write-up of Jack Roberts: https://onlinelibrary.wiley.com/doi/abs/10.1002/anie.201612152
Oh dear. Thanks for catching that, Harold! Fixed.
Hey! Mistake is still there, just so you know.
Thanks for all you posts though! They’ve been a huge help!
Fixed it (finally). thanks for nagging me!