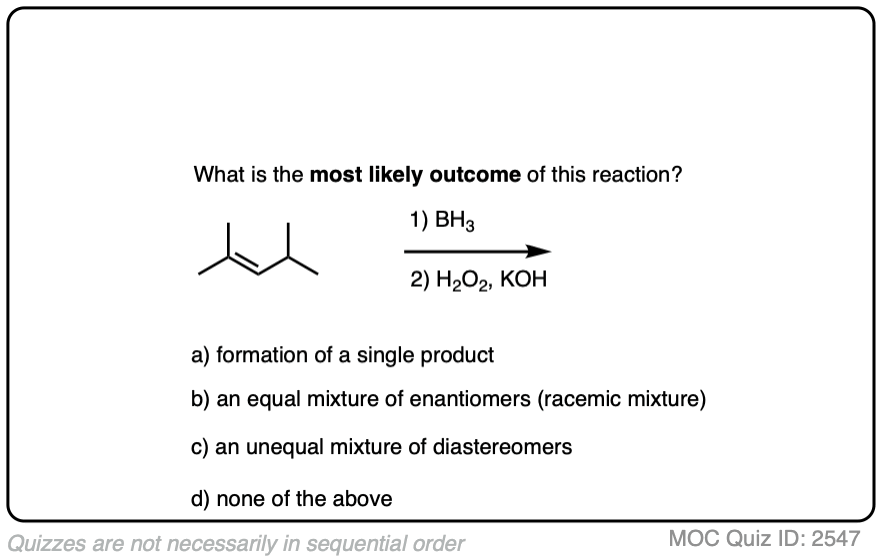
Alkene Reactions
Hydroboration Oxidation of Alkenes
Last updated: February 6th, 2025 |
Hydroboration Oxidation of Alkenes
In this article we cover one of the most important methods for forming alcohols from alkenes, hydroboration-oxidation.
- Hydroboration is an addition reaction between an alkene (olefin) and a a borane (neutral species containing a B-H bond). In hydroboration, a C-C pi bond is broken, and a C-H bond as well as a C-B bond is formed. Oxidation of the resulting organoborane with hydrogen peroxide (H2O2) then replaces the C-B bond with a C-OH bond.
- Typical reagents for hydroboration include borane (BH3) and its relatives (BH3•THF, B2H6, BH3•OEt2) as well as substituted boranes such as disiamyl borane and 9-BBN.
- The notable outcome of hydroboration-oxidation is formation of an alcohol on the least substituted carbon of the alkene (“anti-Markovnikov” regioselectivity. This contrasts with the “Markovnikov” selectivity for formation of alcohols from alkenes using acid-catalyzed hydration (H3O+) or oxymercuration.
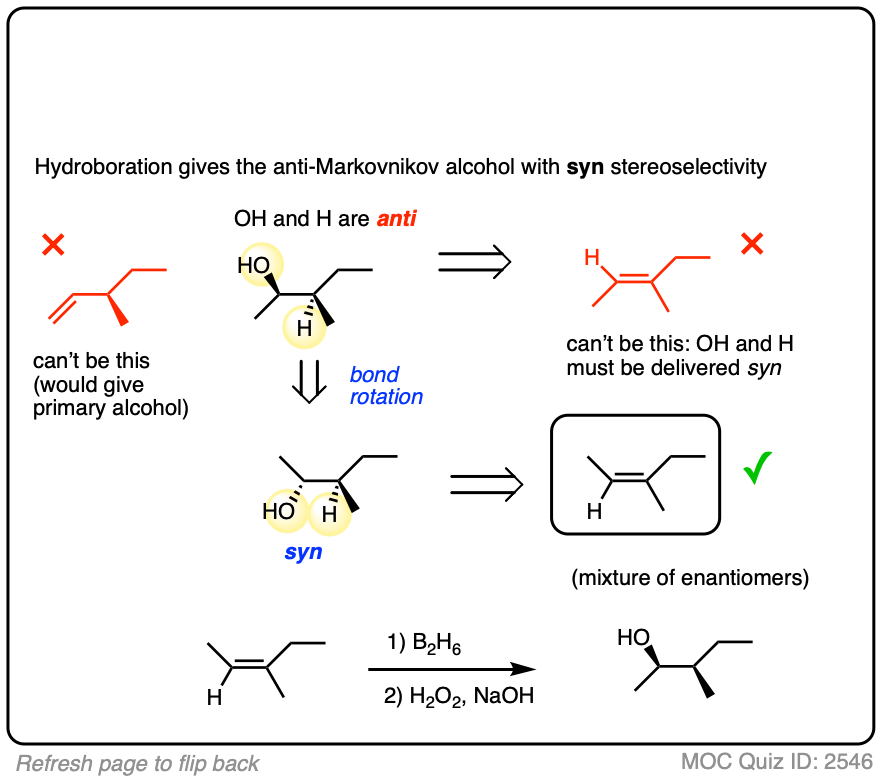
- Hydroboration is stereoselective for syn addition – that is,the H and B are delivered to the same face of the alkene. Oxidation of the C-B bond occurs with complete retention of stereochemistry.
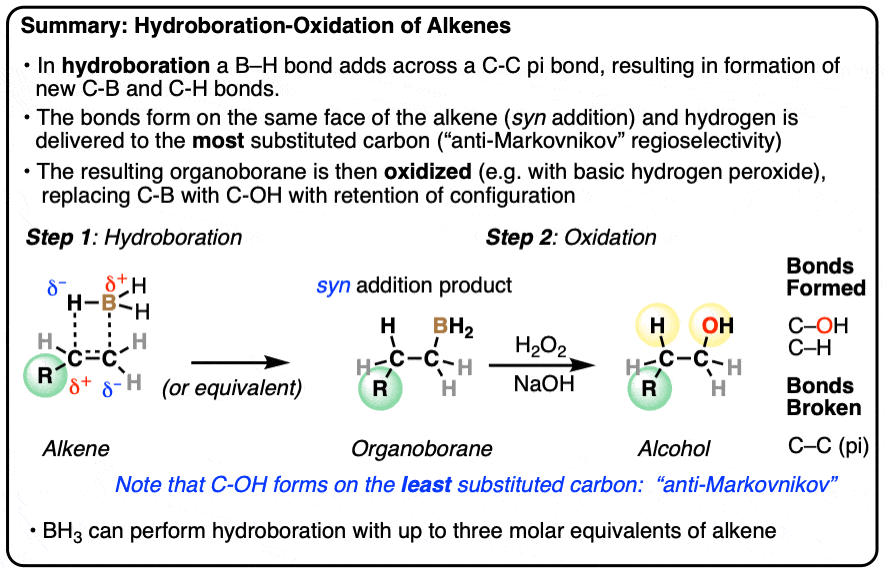
Table of Contents
-
- Hydroboration Oxidation of Alkenes
- Hydroboration-Oxidation is Anti Markovnikov Selective
- Hydroboration is Stereoselective for syn Addition
- The Hydroboration Mechanism
- Mechanism of the Oxidation Step
- Hydroboration is Stereospecific
- Formation of Enantiomers and Diastereomers
- Summary
- Notes
- Quiz Yourself!
- (Advanced) References and Further Reading
1. Hydroboration Oxidation of Alkenes
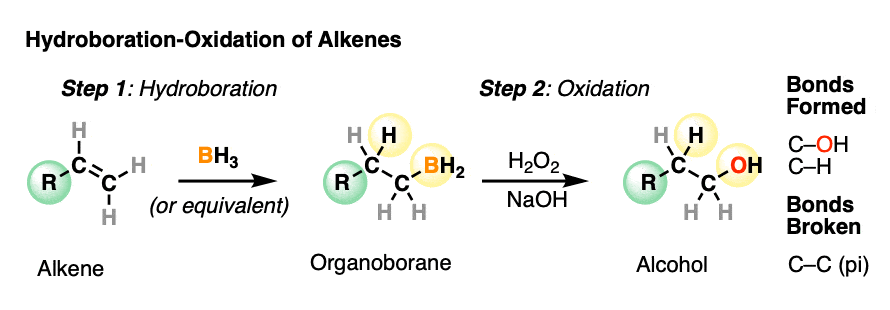
Hydroboration-oxidation is a useful and important method for forming alcohols from alkenes. The reaction involves treatment of an alkene (also known as an olefin) with a borane (a neutral molecule containing a B-H bond). This results in an organoborane compound, which is generally not isolated but instead immediately treated with an oxidant to give the alcohol product.
- In the first step, an alkene is treated with borane (BH3) or any one of similar reagents. This breaks the C-C pi bond and forms a C-H and C-B bond.
- In the second step an oxidant such as H2O2 is added, usually in the presence of base such as NaOH or KOH. A rearrangement then occurs where the C-B bond is broken and a new C-O bond is formed.
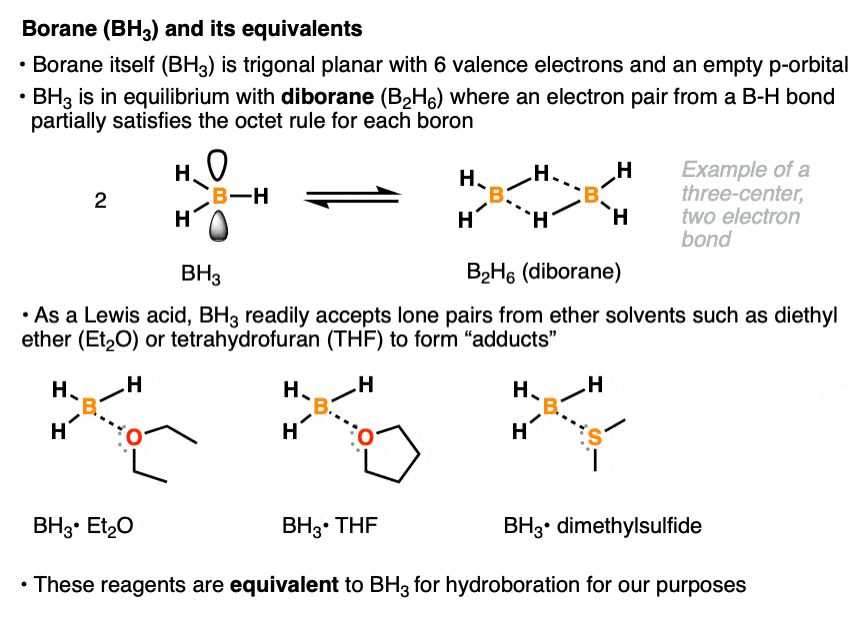
The borane reagent for hydroboration can come in many specific forms. Like all neutral boron species, borane itself (BH3) has six valence electrons and an empty p-orbital. In pure form, it is mostly present as diborane (B2H6) a species where each boron atom partially achieves a full octet of electrons via sharing of a pair of electrons from a B-H bond. [Note 1 ] . With its empty p-orbital, BH3 readily accepts a lone pair of electrons even from weak Lewis bases such as diethyl ether (Et2O), tetrahydrofuran (THF), and dimethyl sulfide (DMS). Various Lewis acid-Lewis base compounds of BH3 (known as adducts) are stable enough to allow for their bottling and use as commercially available reagents, although for our purposes they will all be treated as equivalent to BH3. [Note 2]
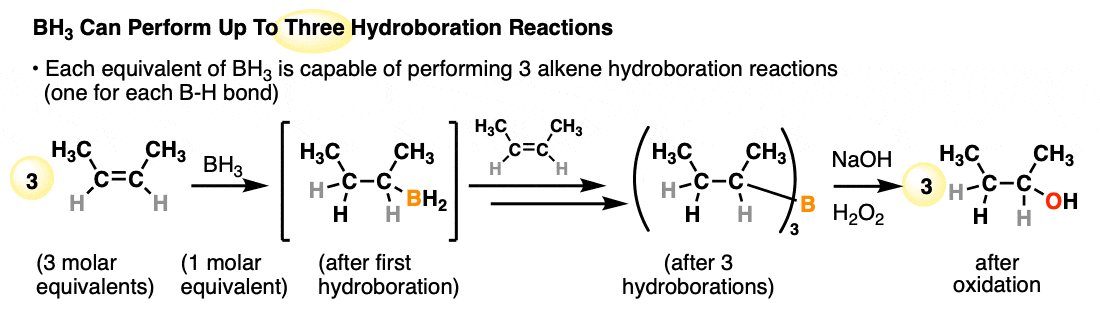
Each available B-H bond of a borane is capable of performing a hydroboration reaction. One molar equivalent of borane (BH3) is therefore capable of performing hydroboration on up to three equivalents of alkene:
There are also many varieties of dialkylborane reagents (R2BH) such as disiamyl borane or 9-BBN that have more steric hindrance around the boron atom and are often used for hydroboration of alkynes. [Note 3] (See post – Hydroboration of Alkynes).
2. Hydroboration Oxidation is Anti-Markovnikov Selective
Hydroboration is an example of an addition reaction, where a C-C pi bond on an alkene is broken, and two new single bonds to C are formed.
Any time addition reactions occur on unsymmetrical alkenes there arises the possibility for forming at least two constitutional isomers.
When a reaction demonstrates a strong preference for the formation of one constitutional isomer over another, it is said to be regioselective.
We previously saw, for example, that addition of acids such as HCl to alkenes tends to give the product where the C-Cl bond forms on the more substituted carbon of the alkene (i.e. the carbon attached to the most carbons and fewest hydrogens) and the C-H bond forms on the less substituted carbon of the alkene (i.e. the carbon attached to the fewest carbons and most hydrogens). Addition of HCl to alkenes, as well as many other reactions are said to have “Markovnikov” regioselectivity. [See article: Markovnikov Addition of HCl To Alkenes]
Hydroboration swings the other way. Hydroboration reactions generally result in the new C-OH bond being formed on the less substituted carbon of the alkene with the C-H bond being formed on the more substituted carbon. This is called, as you might expect, “anti-Markovnikov” regioselectivity.
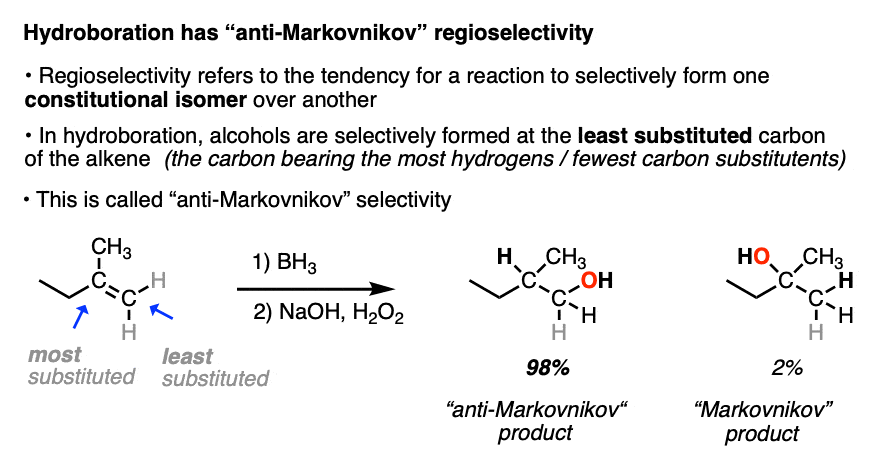
When the carbons of the alkene are bonded to the same numbers of hydrogens, hydroboration with BH3 is much less regioselective. [Note 4 ]
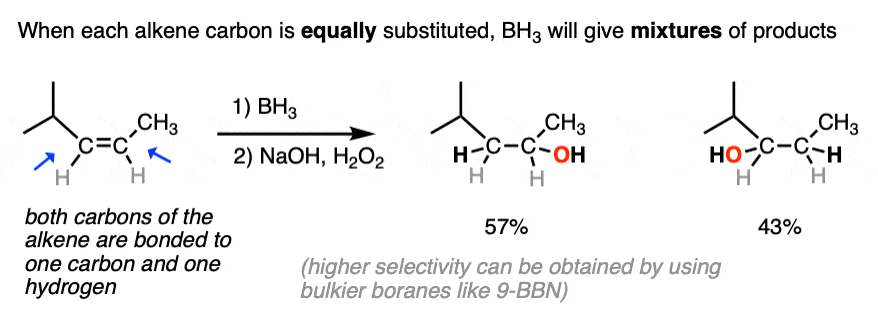
The anti-Markovnikov regioselectivity of hydroboration complements nicely with the Markovnikov selectivity for the formation of alcohols through acid-catalyzed hydration or oxymercuration:
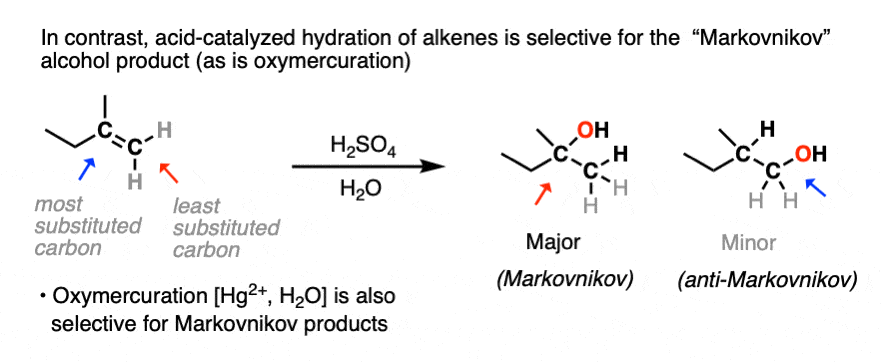
You may recall, however, that acid-catalyzed hydration can lead to carbocation rearrangements. [See article: Rearrangements in Alkene Addition Reactions] Oxymercuration is an alternative route for the formation of Markovnikov alcohol products that don’t have problems with rearrangements.
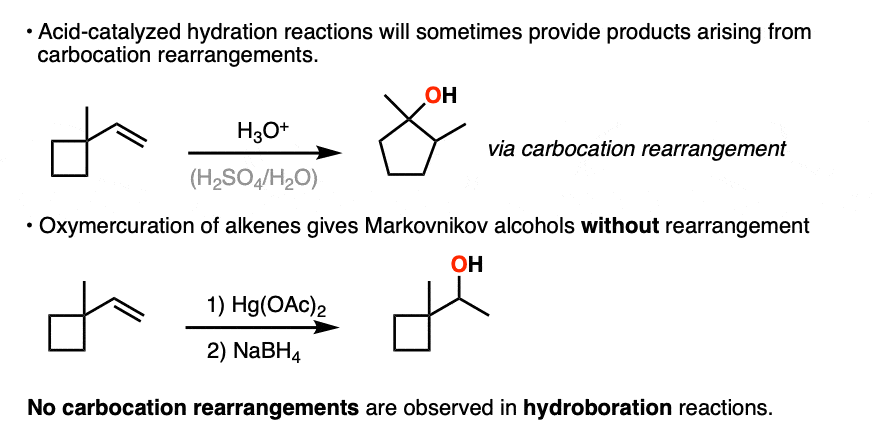
The hydride and alkyl shifts that can happen during acid-catalyzed hydration reactions are never observed during hydroboration reactions, which are a good clue that hydroboration doesn’t pass through a carbocation intermediate.
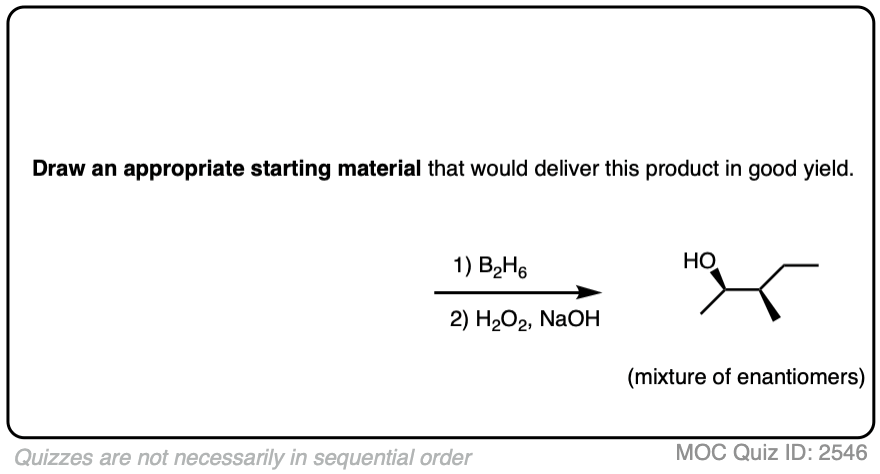
Now for some practice. Knowing that hydroboration gives anti-Markovnikov alcohols, draw the products of these reactions:
 Click to Flip
Click to Flip
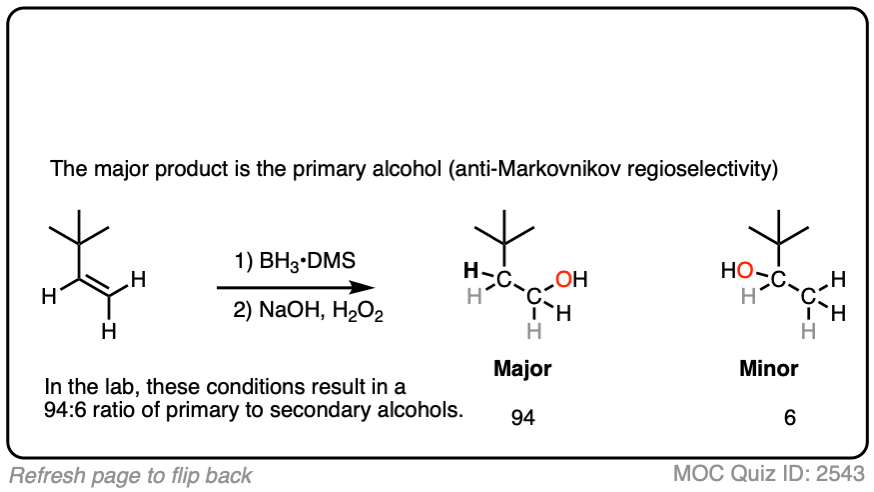
Here’s another one:
 Click to Flip
Click to Flip
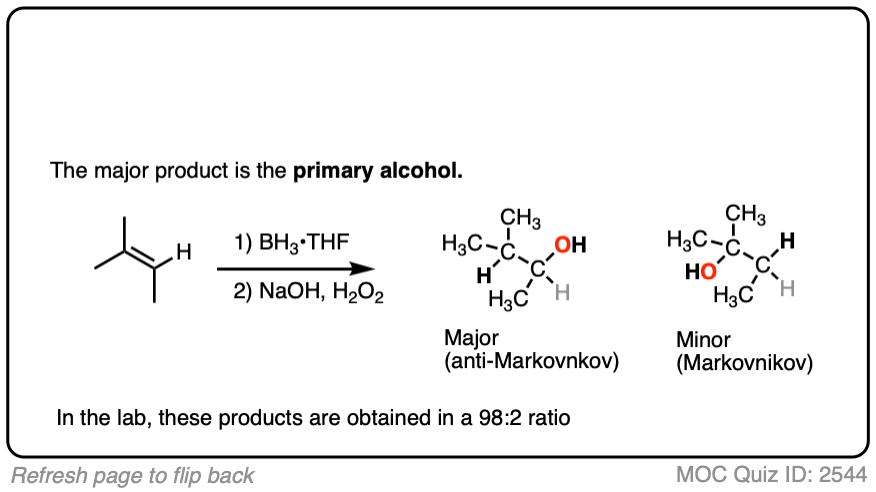
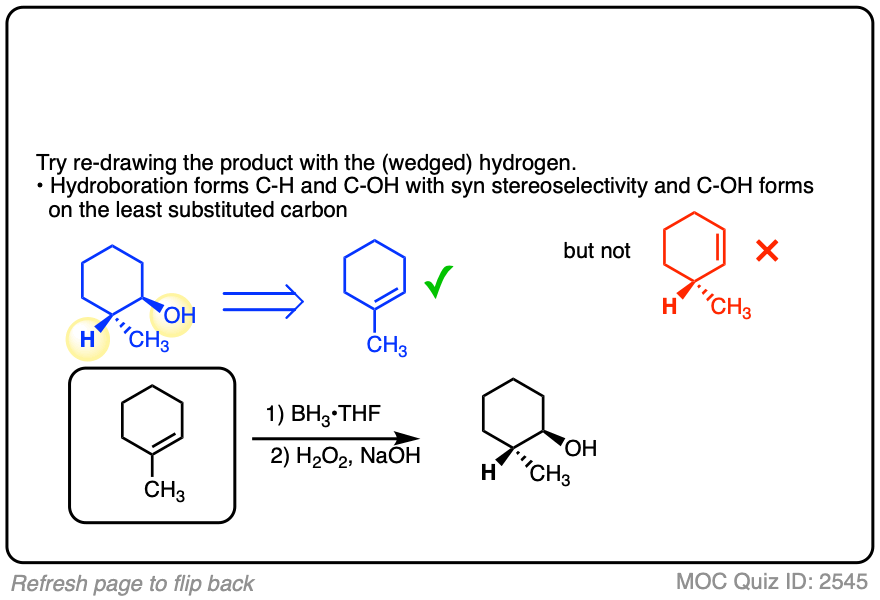
3. Hydroboration-Oxidation is Stereoselective for syn Addition
In addition to being regioselective for formation of anti-Markovnikov alcohols, hydroboration is stereoselective for syn addition products.
Syn addition means that the two new single bonds form on the same face of the alkene. (Other examples of reactions that perform syn addition are catalytic hydrogenation and dihydroxylation).
Addition to 1-methylcyclopentene, for example, produces only the products where C-OH and C-H have added to the same face of the pi bond. No products of anti addition are observed.
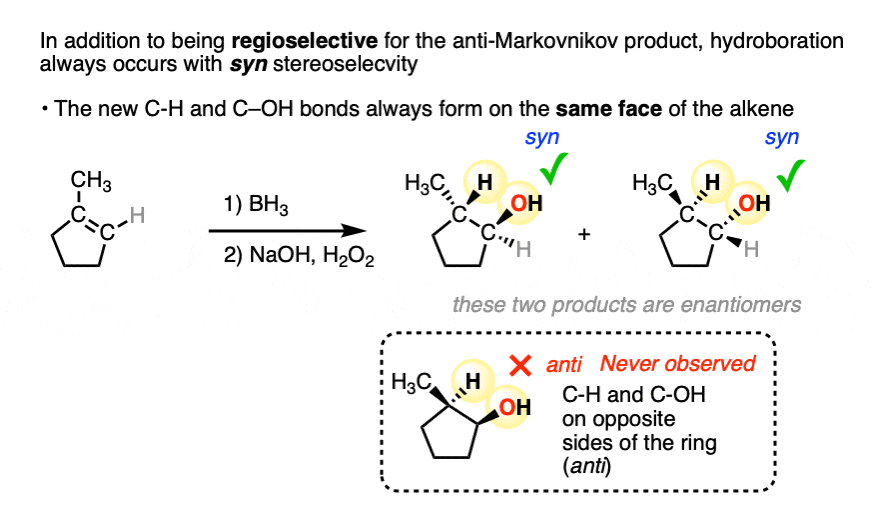
[Note 5]
Try drawing the product of this reaction:
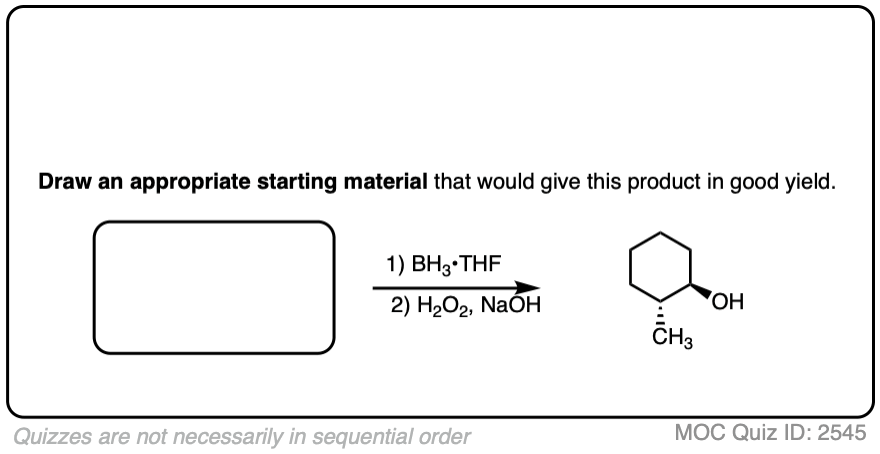 Click to Flip
Click to Flip
Alternatively, try working backwards from the final product. (But be careful! )
 Click to Flip
Click to Flip
4. The Hydroboration Mechanism
OK. So hydroboration is observed to have anti-Markovnikov regioselectivity and is stereoselective for the syn product. It also doesn’t give rearranged products. So how does it work?
Our best theory that fits all the evidence for the mechanism of hydroboration is that it happens via a concerted, cyclic transition state:
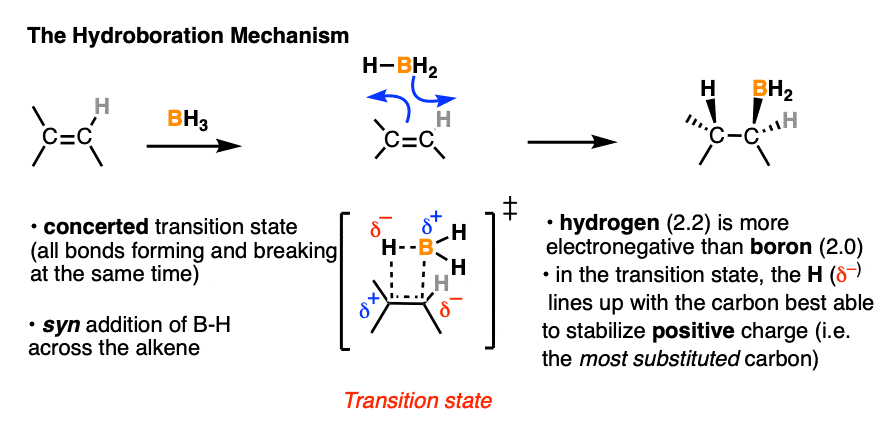
This mechanism accounts for
- the exclusive observation of syn products (C-B and C-H are formed at the same time)
- the lack of any observed carbocation rearrangements (the reaction proceeds in one step, without an intermediate)
- the observed “anti-Markovnikov” selectivity
More on that last point. What’s the origin of this “anti-Markovnikov” selectivity we’ve been discussing, anyway?
Well, in the transition state drawn above, note that the most electronegative (δ– ) atom ends up forming a bond to the carbon best able to stabilize positive charge (i.e. the more substituted carbon) and the most electron-poor atom (δ+ ) ends up bonded to the carbon less able to stabilize positive charge (i.e. the least substituted carbon).
If you stop to think about that for a moment, that explanation doesn’t sound so different from the explanation for Markovnikov selectivity in the addition of HCl (and similar species) to alkenes, now does it? (See post: Markovnikov selectivity]
That is correct. The explanation for “anti-Markovnikov” selectivity in hydroboration is essentially the same as the explanation for “Markovnikov” selectivity in addition of hydrogen halides.

What gives?
- In both cases the more electronegative atom ( δ–) lines up with the more substituted carbon. But the polarity of the hydrogen is different in each case.
- In the B-H bond, the most electronegative atom (δ– ) is hydrogen (2.2) and the least electronegative atom (δ+ ) is boron (2.0). In HCl, the most electronegative atom is Cl (3.16) and the least electronegative atom is H (2.0).
In other words, hydrogen is more electronegative (δ– ) relative to boron (δ+ ) , but less electronegative (δ+ ) relative to halides (δ– ). It just so happens that when H is bonded to boron, it is the more electronegative atom and will therefore line up with the carbon best able to stabilize positive charge (i.e. the more substituted carbon).
So the observed regioselectivity isn’t so “weird” after all.
As for the oxidation step…. OK, that is a little bit weird.
5. Mechanism of the Oxidation Step
After hydroboration is complete, the resulting organoborane needs to be treated with an oxidant like H2O2 to obtain an alcohol.
(Probably best not to try to isolate the organoborane unless you are familiar with inert atmosphere (Schlenk-line) techniques, as organoboranes have a tendency to spontaneously combust in air. Very pretty green flame, though).
This is usually done under basic conditions.
(First, the conjugate base of H2O2 is more nucleophilic than H2O2 itself, and secondly, a base like NaOH helps with hydrolysis of the boronic ester that results after rearrangement).
The first thing to happen (after deprotonation of H2O2) is addition of the peroxide anion to boron.
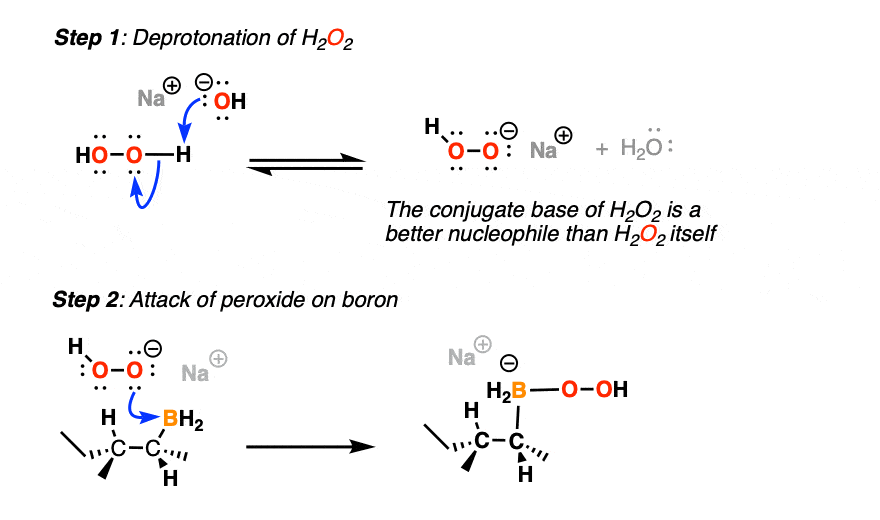
Now comes the fun part. In the next step, the pair of electrons in the C-B bond migrates over to oxygen (form C-O, break C-B). The O-O bond breaks, liberating the hydroxide ion HO(-) as a leaving group.
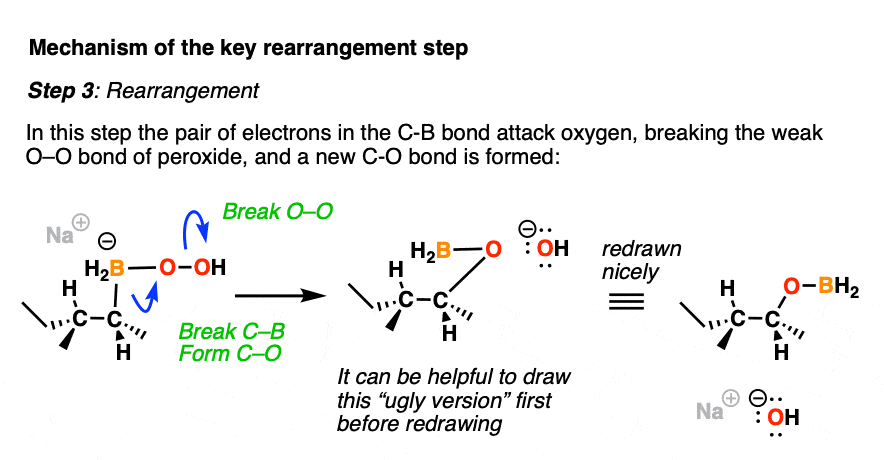
This probably looks weird. It might help to think of the C-B bond as as a nucleophile, attacking the electrophilic oxygen and liberating a hydroxide leaving group.
Normally, oxygen is not the type of atom that undergoes attack by a nucleophile, but the oxygen-oxygen bond is quite weak (roughly 45 kcal/mol); blame it on all those electron pairs repelling each other! These types of rearrangements are not uncommon in peroxide chemistry. [A similar rearrangement is found in a reaction we often encounter in Org 2 known as the Baeyer-Villiger Oxidation]
For accounting purposes, it can be helpful to draw the ugly version first, just keeping track of the bonds that form and break.
After migration, the resulting compound (a “boronic ester”) then undergoes hydrolysis by hydroxide ion to eventually form boric acid and the alcohol.
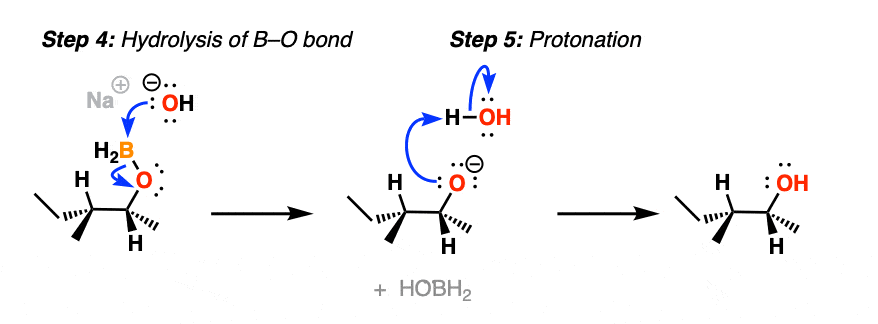
Note that stereochemistry is conserved throughout the rearrangement process. The C-B bond is converted to C-O with perfect fidelity.
6. Hydroboration is Stereospecific
Hydroboration is an example of a stereospecific reaction (a feature it shares with several other reactions of alkenes, including dihydroxylation, epoxidation, and cyclopropanation among others).
Sometimes the word “stereospecific” is used to describe reactions that are super-dee-duper 100% selective for a certain outcome (e.g. saying hydroboration is inherently “stereospecific” for the syn product due to its concerted mechanism).
A more IUPAC-approved definition of stereospecific refers to the fact that you can take two stereoisomeric reactants that differ only in the configuration at one carbon (e.g. the (E) and (Z) alkenes, below) and the products themselves will also be stereoisomers.
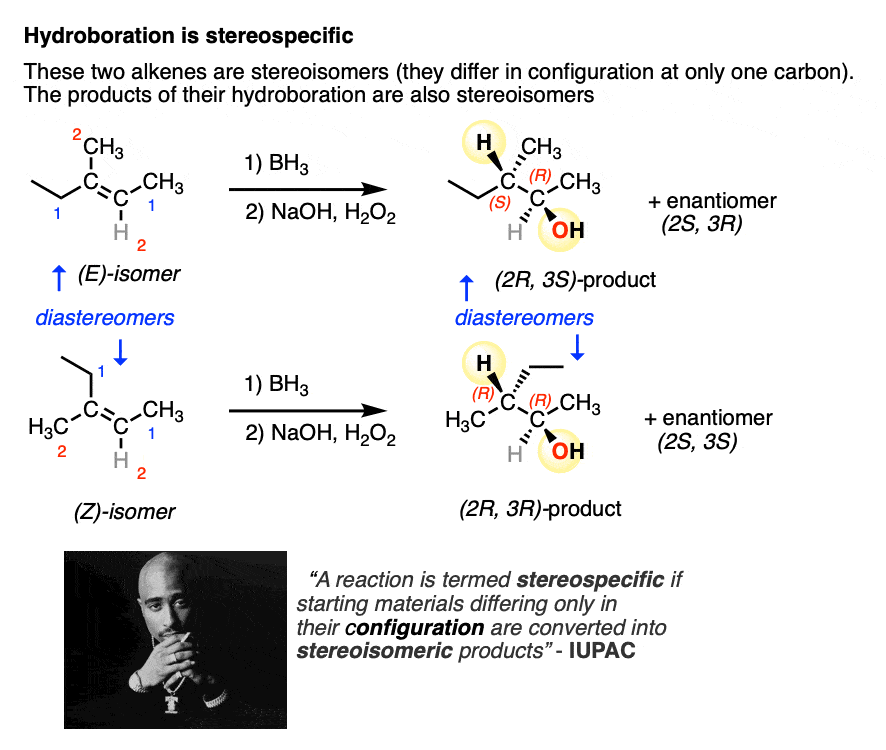
So, in addition to being 100% selective for the syn product, hydroboration also fits this definition of stereospecificity.
7. Formation of Enantiomers and Diastereomers
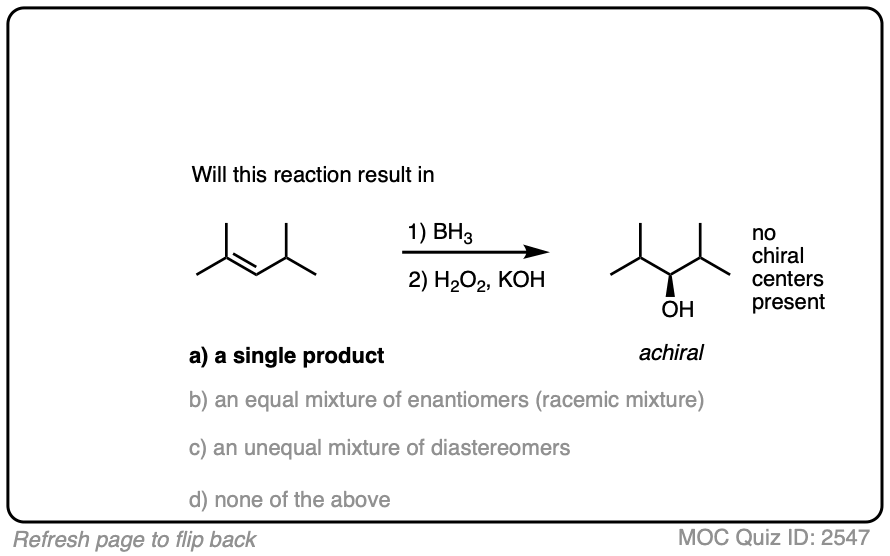
Being planar, alkenes can undergo addition reactions from either face of the pi bond.
Depending on the structure of the alkene, this may lead to the creation of a new chiral center. In the absence of any chiral influences (e.g. by starting with an optically enriched starting material or using an optically enriched reagent) formation of a new chiral center will result in a racemic mixture. (Matt calls this “preservation of optical inactivity”. )
If you’re already familiar with solving “enantiomers, diastereomers, or the same” problems, you should be fine. [See article: Enantiomers, Diastereomers or The Same?] So long as you are able to successfully apply the proper pattern of regioselectivity (anti-Markovnikov) and stereoselectivity (syn) on your starting alkene, it’s much better to just draw out the products and analyze their relationships from there rather than try to memorize all the specific cases that could come up.
 Click to Flip
Click to Flip
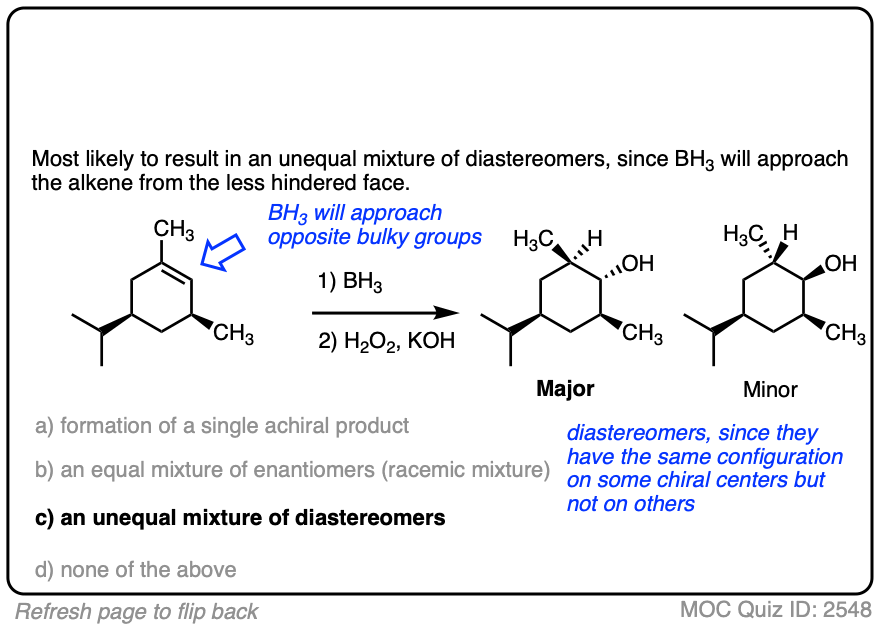
One good thing to keep in mind, however, is that the presence of a pre-existing chiral center will render the two faces of the alkene non equivalent and lead to the formation of a mixture of diastereomers. (with each diastereomer being present as a racemic mixture).
Diasteromers will generally not be formed in equal amounts since one approach of the alkene is often favored over the other due to differing steric hindrance encountered by the electrophile as it approaches the alkene.
A classic example is the hydroboration of norbornene, a vaguely tent-shaped molecule.
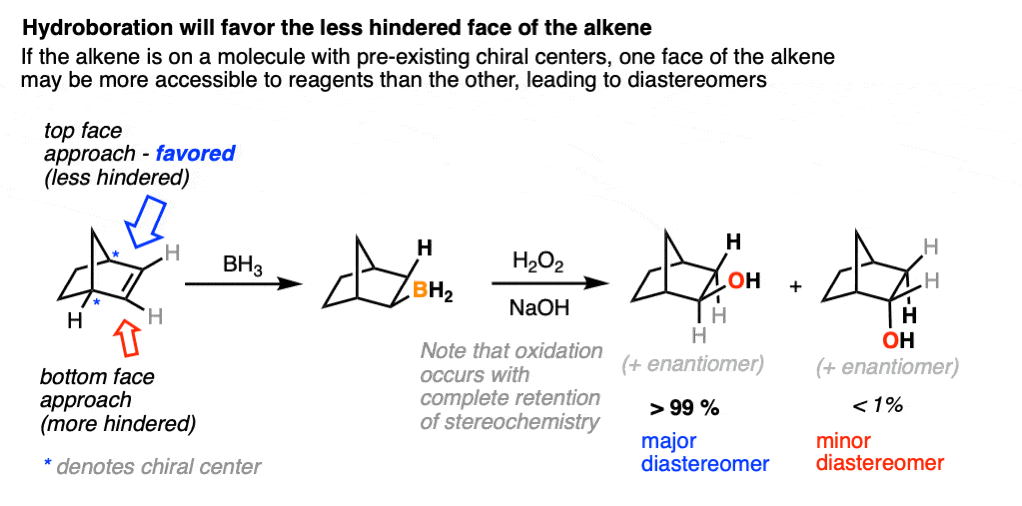
Hydroboration occurs exclusively on the outside of the tent to give only one product (as a mixture of enantiomers). The other diastereomer is formed in such tiny quantities that it is not generally observed.
Here is another example. See if you can predict the major product.
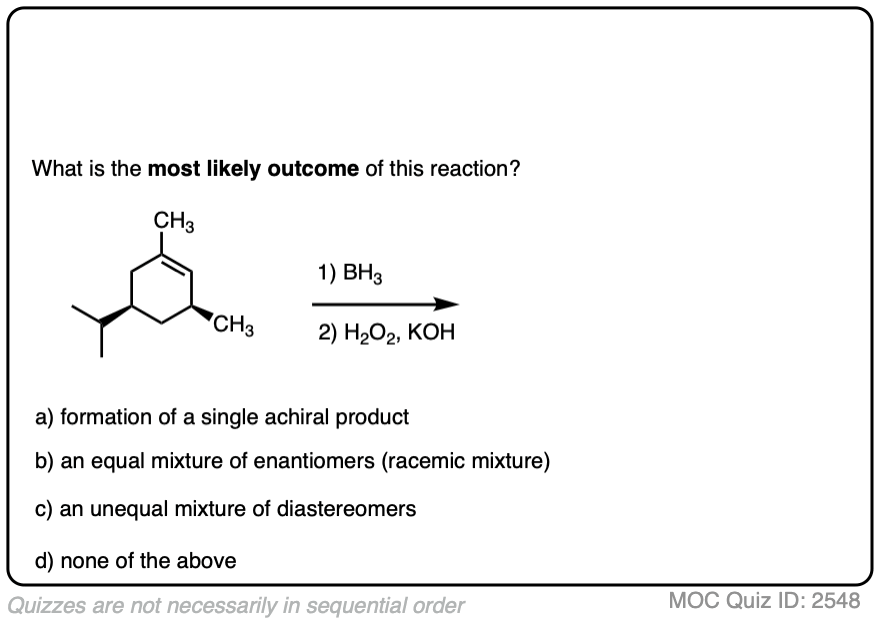 Click to Flip
Click to Flip
8. Summary
- Hydroboration-oxidation is an addition reaction that results in the formation of alcohols.
- A classic reagent for hydroboration is borane (BH3) and related reagents (e.g. BH3•THF) which can perform hydroborations on up to three molar equivalents of alkene. Substituted organoboranes such as disiamyl borane and 9-BBN have a single B-H bond and can be useful for the hydroboration of alkynes.
- In hydroboration-oxidation a new C-OH bond is generally formed on the less substituted carbon of the alkene. This is referred to as “anti-Markovnikov” regioselectivity. Hydroboration is stereoselective, giving exclusively syn addition; the C-H and C-B bonds are formed on the same face of the alkene. Oxidation with hydrogen peroxide (H2O2) occurs with complete retention of stereochemistry on the C-B bond.
- The key step of the oxidation reaction is the shifting of a pair of electrons in the C-B bond to form a new C-O bond, with breakage of the relatively weak (45 kcal/mol) O-O bond. The resulting boronic ester is then hydrolyzed with base to give an alcohol.
Notes
This is an updated version of two older articles (Hydroboration-Oxidation of Alkenes, and Hydroboration of Alkenes – The Mechanism) which have been combined into one larger article.
Note 1. This is an example of a three-center, two-electron bond, where two bonding electrons are shared between two atoms. The bridging B-H bonds are significantly longer (1.31 Å) than the non-bridging B-H bonds. (1.19 Å)
Note 2. The decision to use one reagent or the other (e.g. BH3•OEt2 vs BH3•SMe2 vs B2H6) is often based more on factors like price, availability, shelf life, and convenience rather than their reactivity profiles.
Note 3. Structures of 9-BBN and disiamylborane.
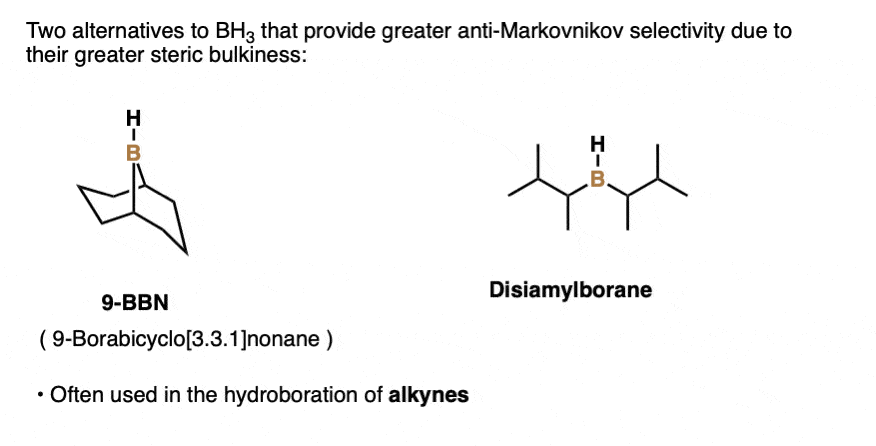
Note 4. This 57:43 ratio can be improved significantly, however, by using bulkier reagents for hydroboration (such as 9-BBN or disiamylborane, above). With disiamylborane the ratio is 97:3 and with 9-BBN it is over 99:1. [Page 12 in the PDF of Ref]
Note 5. Note that while syn and cis are sometimes used interchangeably, they are actually distinct. cis- and trans- refer to the relative orientations of two groups about a multiple bond or ring where rotation is impossible (e.g. cis and trans alkenes) whereas syn and anti is defined by the dihedral angle between two bonds A-B and C-D in a four-atom system A-B-C-D. Dihedral angles, and therefore syn and anti relationships can change with bond rotation. [See article – Staggered vs. Eclipsed Conformations of Ethane] When we say “syn” addition we are referring to the relative orientation of the two new bonds in the transition state.
Note 6. In olden days, BH3 was used for the reduction of the carbonyl group (such as aldehydes to alcohols). Hydroboration was serendipitously discovered during one of these reactions by H.C.Brown’s co-worker Subba Rao, who noticed that reduction of an ester consumed “extra” BH3 beyond the two equivalents expected per ester.

Quiz Yourself!
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.

Become a MOC member to see the clickable quiz with answers on the back.
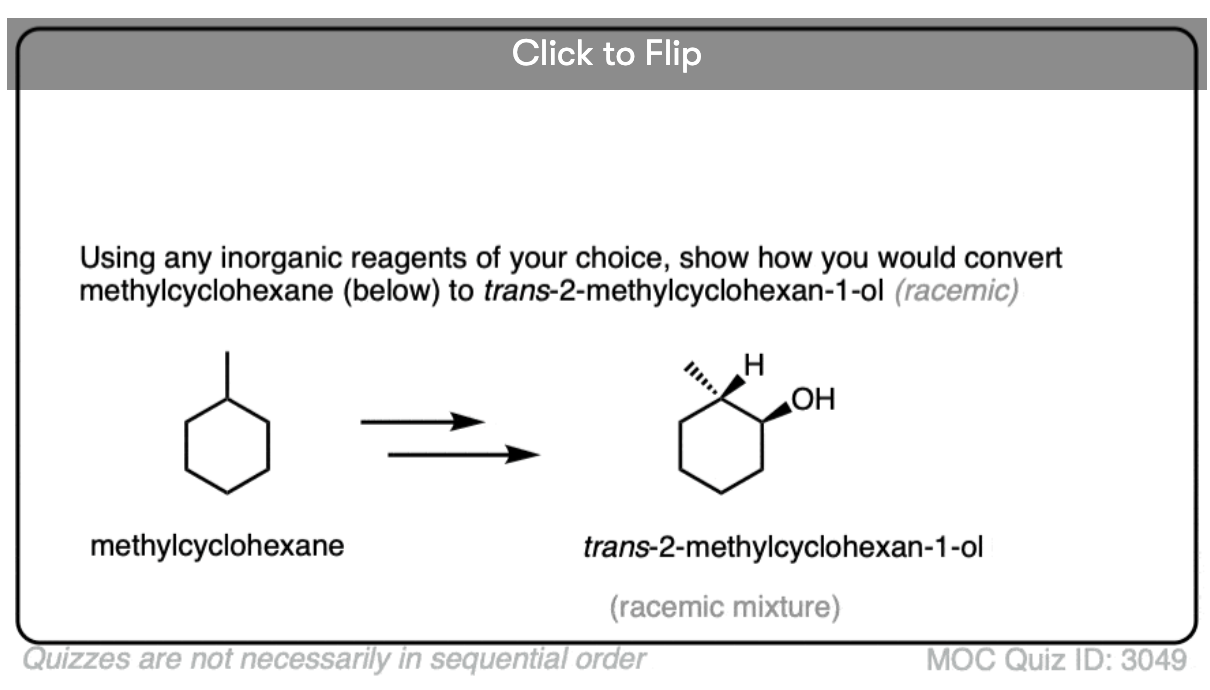
Become a MOC member to see the clickable quiz with answers on the back.
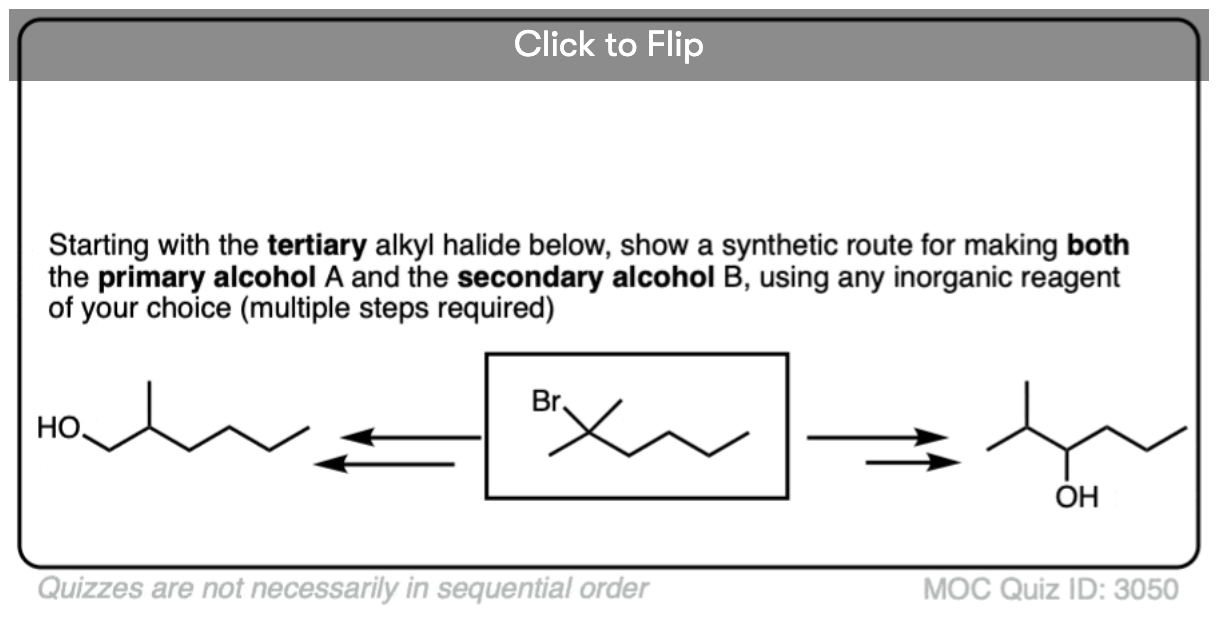
Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading
One of the best overall references that covers the basics is Organic Syntheses via Boranes (Wiley, 1975) by H. C. Brown. It’s available for 1 hour loans on archive.org . Link.
H.C. Brown’s Nobel Lecture is also well worth reading for the story of how his research program on boron chemistry evolved. Some of the earliest techniques for working with air-sensitive reagents were developed in Brown’s lab.
- A STEREOSPECIFIC CIS HYDRATION OF THE DOUBLE BOND IN CYCLIC DERIVATIVES
Herbert C. Brown and George Zweifel
Journal of the American Chemical Society 1959 81 (1), 247-247
DOI: 10.1021/ja01510a059 - (−)-ISOPINOCAMPHEOL
George Zweifel and H.C. Brown
Org. Synth. 1972, 52, 59
DOI: 10.15227/orgsyn.052.0059 - XXIV. Directive Effects in the Hydroboration of Some Substituted Styrenes
Herbert C. Brown and Richard L. Sharp
Journal of the American Chemical Society 1966 88 (24), 5851-5854
DOI: 10.1021/ja00976a029
A very nice Physical Organic study of the hydroboration of styrenes, involving a Hammett plot (a classic tool in physical organic chemistry) to determine a relationship between the stereochemistry of the reaction and the electron density of the alkene. - A SIMPLE AND CONVENIENT METHOD FOR THE OXIDATION OF ORGANOBORANES USING SODIUM PERBORATE: (+)-ISOPINOCAMPHEOL
George W. Kabalka, John T. Maddox, Timothy Shoup, and Karla R. Bowers
Organic Syntheses, Coll. Vol. 9, p.522 (1998); Vol. 73, p.116 (1996)
DOI: 15227/orgsyn.073.0116
A Hydroboration-oxidation procedure where the oxidizing agent is sodium perborate, a cheap, safe, easily handled oxidant commonly used in laundry detergents. - Hydroboration with Pyridine Borane at Room Temperature
Julia M. Clay and and Edwin Vedejs
Journal of the American Chemical Society 2005 127 (16), 5766-5767
DOI: 1021/ja043743j
A modern method for doing hydroboration at room temperature using pyridine-borane, which usually requires heating to 75-100 °C to liberate the borane.
In tent shaped alkene molecule why bottom face approach more hindered .can you explain more
In solution, the molecule can be approached by a reactant from every direction (360 degrees). If the molecule were perfectly flat, then half (180 degrees) of the molecule would be approachable from the top and the other half (180 degrees) from the bottom.
In the tent shaped molecule the top is approachable from more like 270 degrees and the bottom something like 90 degrees. (This is an oversimplification, but it will do for our purposes).
Statistically, due to the shape of the molecule, it is more likely for reactants to approach from the top face.
Why are most substituted carbons more positive in charge. Seems counter intuitive since carbons are electron donating, so the carbon with more carbons attached to it should have a partial negative charge since there is a higher electron concentration right? Why is it the other way around.
I tell my students Boron is just a whiner, never happy, always complaining; “I want an Octet, everyone else has an octet, except H but it doesn’t even want an octet. Wait now I have a Negative Charge, I don’t want a Negative Charge…”. Then I say Al is a more aggressive whiner, its just angry! “I WANT AN OCTET NOW!”
If interested see slides 10 and 11 of my animation about reductions with NaBH4 and LiAlH4. Fun stuff…
https://docs.google.com/presentation/d/1u1YSKJN2D9nRz0BkG18prvmlSnURum1fOsMoafurJ5U/edit#slide=id.p4
Nice! As the parent of a toddler, it resonates.
Hello!
I thought that electronegativity was a constant for an atom. You indicate H=2.2, Cl=3.2, Br=3.0.
OK, this is the value given in the periodic table.
But further down, you indicate that in BH3, boron is less electronegative than H: if electronegativity is a constant, I don’t understand anymore? Why is that?
Br is bromine (electronegativity 3.0). The symbol for boron is B (electronegativity 2.0)
In the transition state, the boron should have a partial negative charge (not positive). The B in borane is electrophilic with only 6 valence electrons, and so when the alkene forms a bond to B, it gains a partial negative charge. Hence, at the end of the other double bond, the carbon bears a partial positive charge. Don’t use the electronegativity analogy here, but rather the formal charge.
The partial positive charge reflects the (slight) polarization of the B-H bond, and in turn helps to rationalize the anti-Markovnikov regioselectivity.
Do you have a sorted progression of these blog posts? I love them! It’s just I wish there was a way I could look at topics and then follow the sequence instead of just looking up specific subjects and then following the next blog post.
Yes – check out the new homepage at http://www.masterorganicchemistry.com !
James…..You are just awesome…!!
I am an IIT JEE aspirant and I can’t explain it to you how much it is helping me….GREAT JOB…
Glad it helps, and good luck on your exam!
As we know HB oxidation used for the production of ketone and aldehyde,… why not u give as an ex for those?
Hydroboration of alkenes would not produce ketones or aldehydes directly; you are referring to hydroboration of alkynes, which is covered here. https://www.masterorganicchemistry.com/2013/05/14/hydroboration-and-oxymercuration-of-alkynes/
Hey, why do you say that the partial charge on boron of BH3 is positive?
It should be negative!!
Compare the electronegativities of B and H and tell me where the dipole should be in the B-H bond?
What would have been the product of the oxidation after hydroboration if H2O2 is replaced by D2O2?
Good question! The answer is… R-OD. But it you use a protic solvent like H2O in the workup, the deuterium will exchange for hydrogen, and you’ll end up with R-OH !
On the other hand if you use BD3 instead of BH3, you will form a C-D bond in the product.
OH- is attacking in 4th step and substituting the stronger base RO-. But leaving tendency of stronger base is low as we have seen that -OH group can not be substituted by halogen from alcohol and for that we use haloacids.
Substituting RO- for HO- is very possible with an excess of reagent. They are of comparable basicity.
you helped me a lot to clear some of my confusions. thank you.
Glad to hear it Visva.
what if both locations are equally substituted? Such as in trans-4,4-dimethylpent-2-ene Where will the OH be placed?
You’ll obtain a mixture of products. There won’t be much, if any, regioselectivity. A 55:45 mixture of products is not a great result.
The link in the fourth paragraph that leads to the review on why H-Cl and H-Br are Markovnikov reactions no longer works.
Fixed. Thank you
http://web.chem.ucla.edu/%7Eharding/IGOC/M/markovnikovs_rule.html
Anti Markonokov regioselectivity of the final product is all right, But is’t the addition of BH3 following the Markonikov rule?
Yes, it essentially is. The carbon best able to support positive charge is interacting with the most electronegative group of the electrophile and vice versa.
Just wanted to say I love what you did in this series of posts, the order at which you presented the addition reactions and the reasons for why we need to look for new mechanisms to explain them. This would really help remembering it.
Couple of questions though:
1) Is there a driving force behind the backside attack of the electron pair in step 3?
2) Why is it that the hydroxide anion attacks the boron in step 4 but not in step 1? Is it a question of concentration ratios?
What does the mechanism look like for hydroboration 1. BH3, THF 2. HOOH, OH-, ROH? If we have an alcohol instead of water in the last step.
You will get a borate ester, B(OR)3 . Which is fine for our purposes.
You are great!! Thank you Dr.James
This post saved my life! Thank you so much!
OK, thank you Nayara!
Excellent!!! This is the post I’ve been looking for the past two years!!
Worth noting that the B-to-O migration step is analogous to the migration step in the Baeyer-Villiger reaction (http://www.organic-chemistry.org/namedreactions/baeyer-villiger-oxidation.shtm). That one’s C-to-O, but it’s a similar idea.
Boron loves those migrations…the way it was described to me, it’s conflicted–it doesn’t know whether it wants to have an octet or neutral formal charge. A source is escaping me at the moment, but Zweifel olefination is similar (B-to-C migration).
Yes! the key step in the BV, as well as in the Beckmann, Curtius, Hoffman, Wolff, and other rearrangements. Notoriously difficult for students, even though it’s only a single arrow to draw.
I tell my students that boron is the petulant teenager of the periodic table. It mopes around the house all day whining about how it doesn’t have enough electrons. So someone comes along and gives it two more – then it can’t stop complaining about this stupid negative charge…