Alkyne Reactions
Hydration and Oxymercuration of Alkynes
Last updated: May 28th, 2026 |
Hydration and Oxymercuration of Alkynes Via Keto-Enol Tautomerism
Alkyne chemistry bears many resemblances to alkene chemistry, but in these first few posts on the subject, the purpose is to illustrate how one seemingly minor change – an extra π bond – can lead to significant differences in chemical behavior.
Previously, we saw that the sp- hybridization of alkynes leads to increased acidity, and the second π bond of alkynes leads to the possibility for partial reduction to either cis or trans alkenes. In this post we’ll see again how the addition of that extra π bond has a very important and surprising consequence.

Table of Contents
- Hydration of Alkenes With Aqueous Acid Gives Alcohols
- Hydration of Alkynes With Aqueous Acid Gives… Ketones??… What?!
- The First Step In The Hydration of Alkynes Is Formation Of An “Enol”
- The “Enol” Is Converted To A Ketone Through A Process Called “Tautomerization”
- Alkynes Can Also Be “Hydrated” via Oxymercuration (HgSO4/H2O)
- Hydroboration Of Alkynes (R2BH) Occurs With Anti-Markovnikov Selectivity, Giving Aldehydes From Terminal Alkynes
- Beware: Depending On The Alkyne, Mixtures Of Products Can Be Obtained
- Notes
- Quiz Yourself!
- (Advanced) References and Further Reading
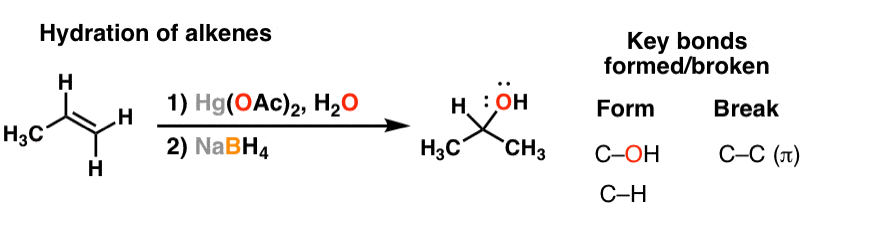
1. Hydration of Alkenes Gives Alcohols
Several posts ago we talked about the hydration of alkenes. This can be done either with aqueous acid, or with mercury and water (“oxymercuration” – see article – Oxymercuration of Alkenes). Looking at the reaction with alkenes, the pattern is fairly straightforward: break a C-C π bond, and form a C-H and C-OH bond. Also recall that the oxygen ends up on the most substituted carbon [“Markovnikov” selectivity].
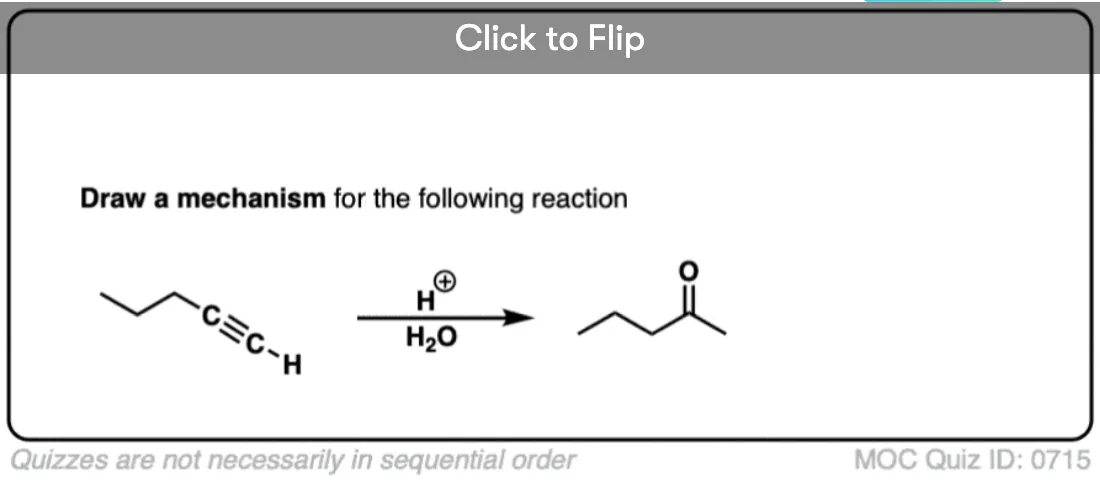
2. Hydration of Alkynes With Aqueous Acid Gives… Ketones??… What?!
So what happens when we try this reaction on alkynes? We might expect to observe the same pattern, right? After all, it’s just a simple addition reaction.
Well… here’s what we actually observe. We get… a ketone !?
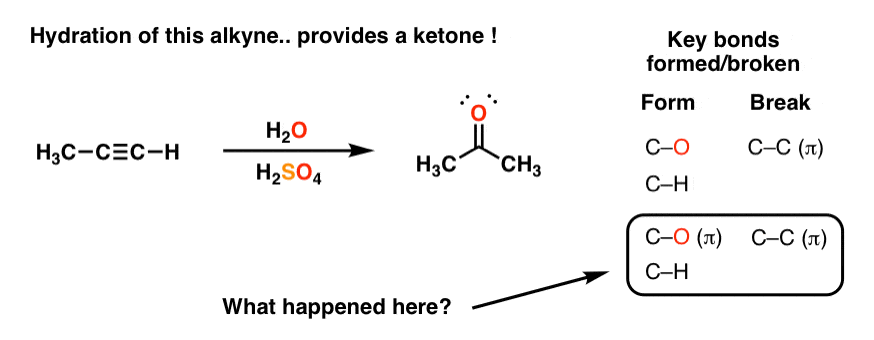
Now what’s going on here? This seems like the type of thing that drives new organic chemistry students around the bend. Just when you think you understand your surroundings, you pick up the most innocuous looking rock, and underneath it find a poisonous snake!
Don’t panic! It’s a new concept in organic chemistry we’ll be exploring here called tautomerism – one that gets much more discussion in Org 2, but it’s not as weird as you initially might think. (See post: Keto-Enol Tautomerism)
Look at the bonds formed and broken. The first set we should understand. Form C-O and form C-H, break C-C π. It’s that next set of bonds formed/broken that are a big surprise.
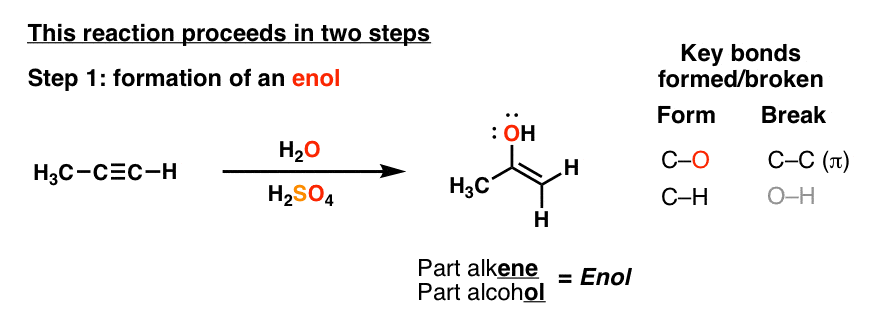
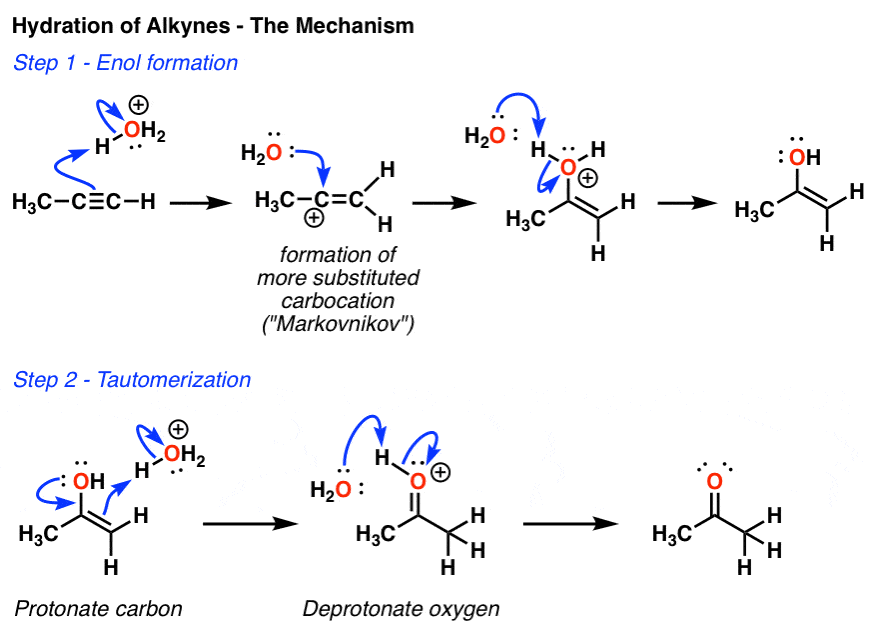
3. The First Step In The Hydration of Alkynes Is Formation Of An “Enol”
If you monitor this reaction closely – one way to do it is in an NMR tube – it’s actually possible to observe the first product of this reaction, which is the one shown below. We call this an “enol”, by the way – kind of like a spork (half spoon half fork) it is part alkene, part alcohol. (See post: Reactions of Enols)
4. The “Enol” Is Converted To A Ketone Through A Process Called “Tautomerization”
Over time, this enol spontaneously converts into the ketone. Note that the two have the same molecular formula – they are constitutional isomers. And they are in equilibrium with each other.
We call these constitutional isomers which interconvert, “tautomers”. This equilibrium generally favors formation of the ketone due to the strong C-O π bond (compared to C-C π).
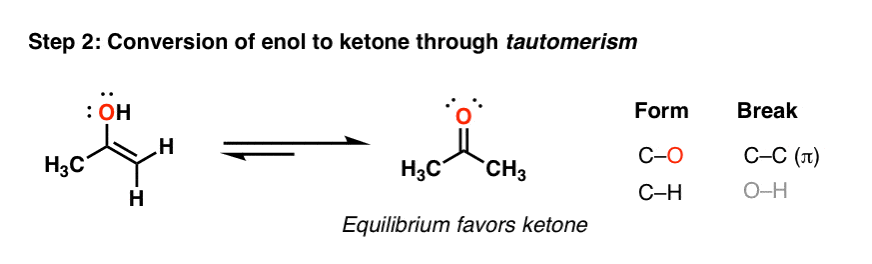
Here’s how the whole process works – arrow by arrow.
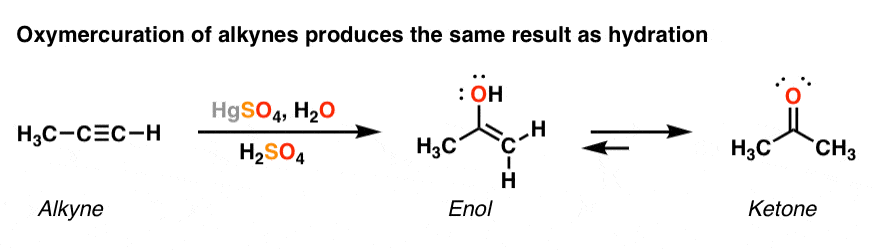
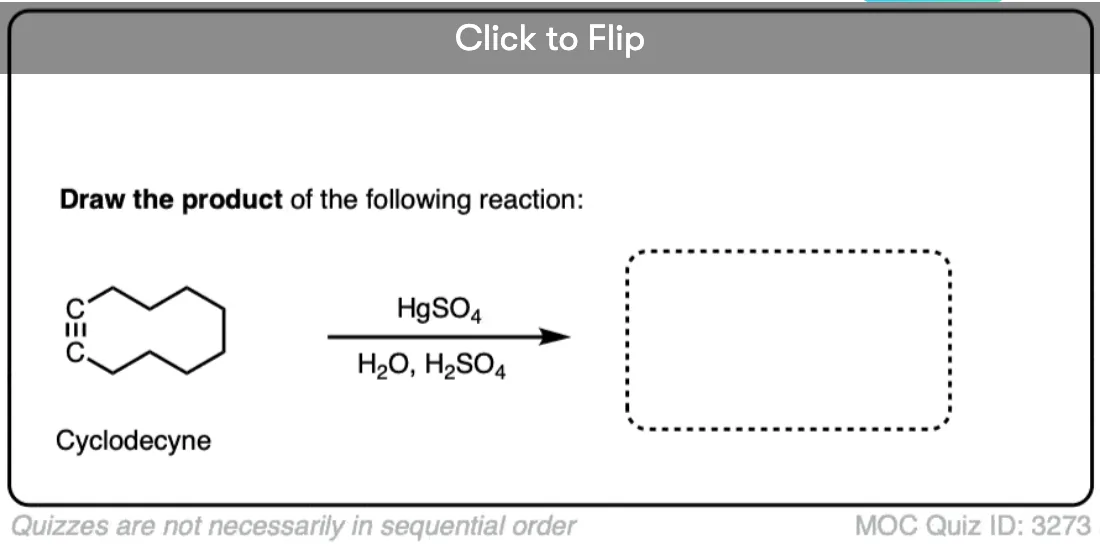
5. Alkynes Can Also Be “Hydrated” via Oxymercuration
Wait – we’re not done! There’s another way to “hydrate” alkynes, just like there was with alkenes. We can also perform the same reaction with mercury, water and strong acid [sulfuric acid, H2SO4 is the usual acid of choice]. For interesting reasons we wont get into at the moment, sodium borohydride (NaBH4)is not generally used for removal of the mercury with alkynes; it is sufficient to merely have water and acid present.
6. Hydroboration Of Alkynes Occurs With “Anti-Markovnikov Selectivity”, Giving Aldehydes From Terminal Alkynes
There’s also hydroboration. Remember how hydroboration-oxidation of alkenes with BH3 and H2O2 gives us “anti-Markovnikov” hydration of alkenes? (See post: Hydroboration of Alkenes)
Likewise, we can use the same reaction to perform “anti-Markovnikov” hydroboration of alkynes.
Just as in the cases above, we initially obtain an enol. However, under the reaction conditions, keto-enol tautomerism results in formation of the aldehyde. (For more, see article: Hydroboration of Alkynes With R2BH)
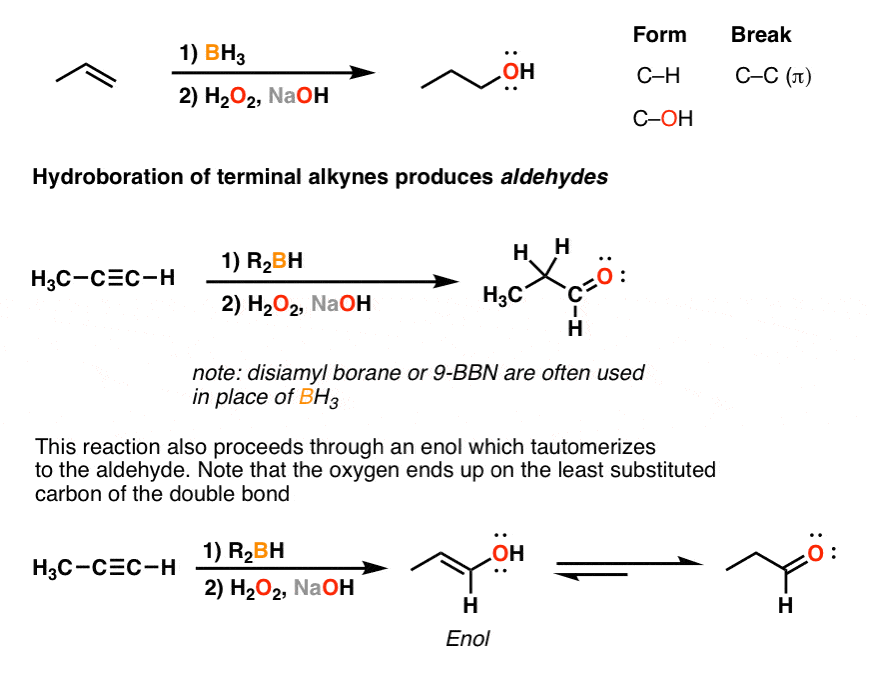
Bottom line here: if we start with a “terminal” alkyne, that is an alkyne where one of the carbons is attached directly to H – then we will obtain ketones with H3O+/H2SO4 or via oxymercuration, and aldehydes via hydroboration.
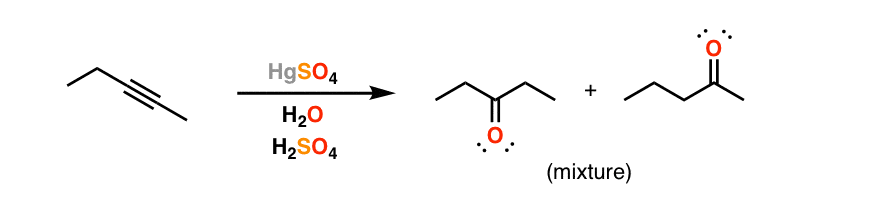
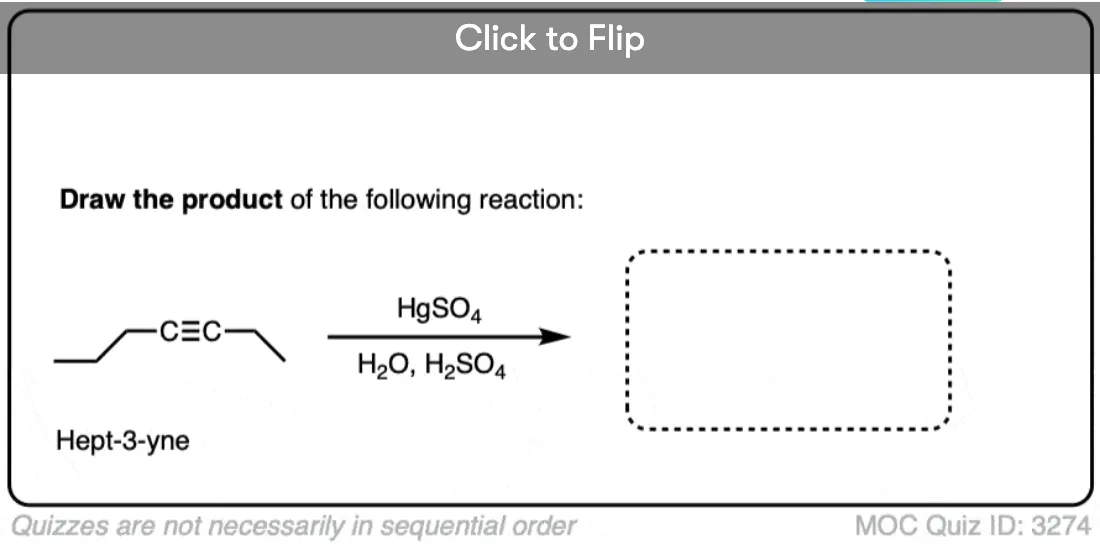
7. Depending On The Alkyne, Mixtures Of Products Can Be Obtained
One final note: if we use an alkyne where both ends are directly attached to carbon, we will obtain a mixture of products. That’s just “Markovnikov’s rule” – remember that if each carbon in the multiple bond is attached to an identical number of hydrogens, then we can’t determine which is the “most substituted” for our purposes. Like in this example.
Next Post: Alkyne Reaction Patterns – The Carbocation Pathway
Notes
Note 1. Note: while BH3 is sometimes written for this, it’s not strictly correct to do so. Why? Double addition (see ref)
Instead often use sterically hindered boranes, such as disiamyl borane or 9-BBN that both increase the proportion of addition to the less substituted carbon and also prevent a second hydroboration reaction.
Quiz Yourself!
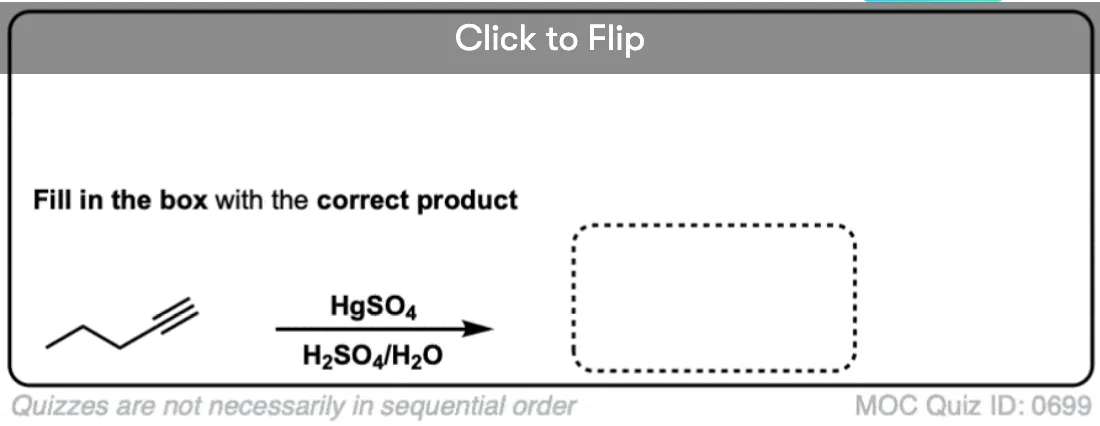
Become a MOC member to see the clickable quiz with answers on the back.
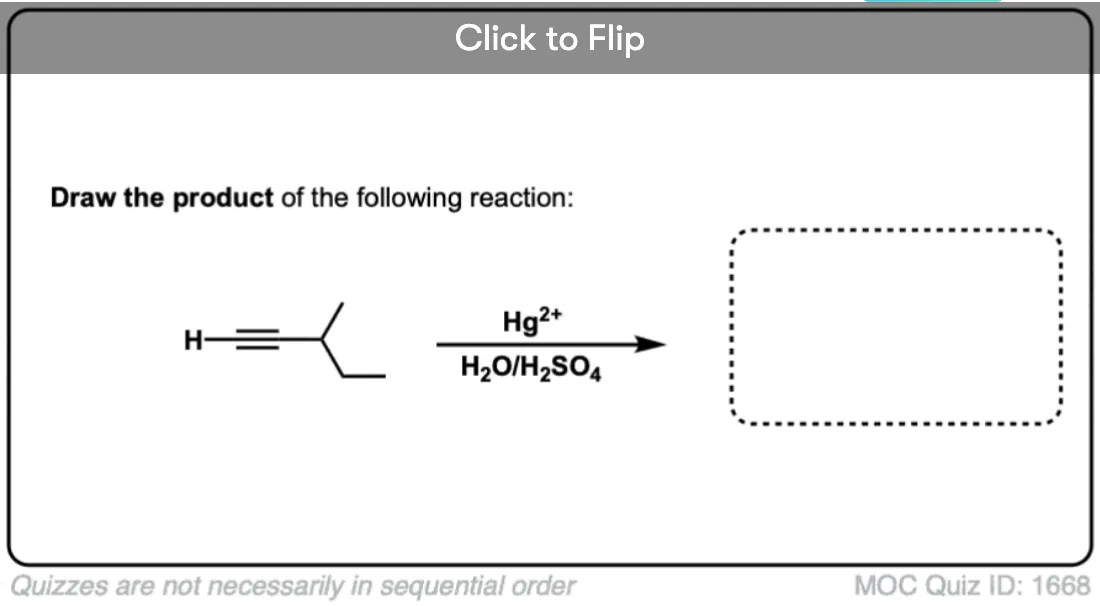
Become a MOC member to see the clickable quiz with answers on the back.
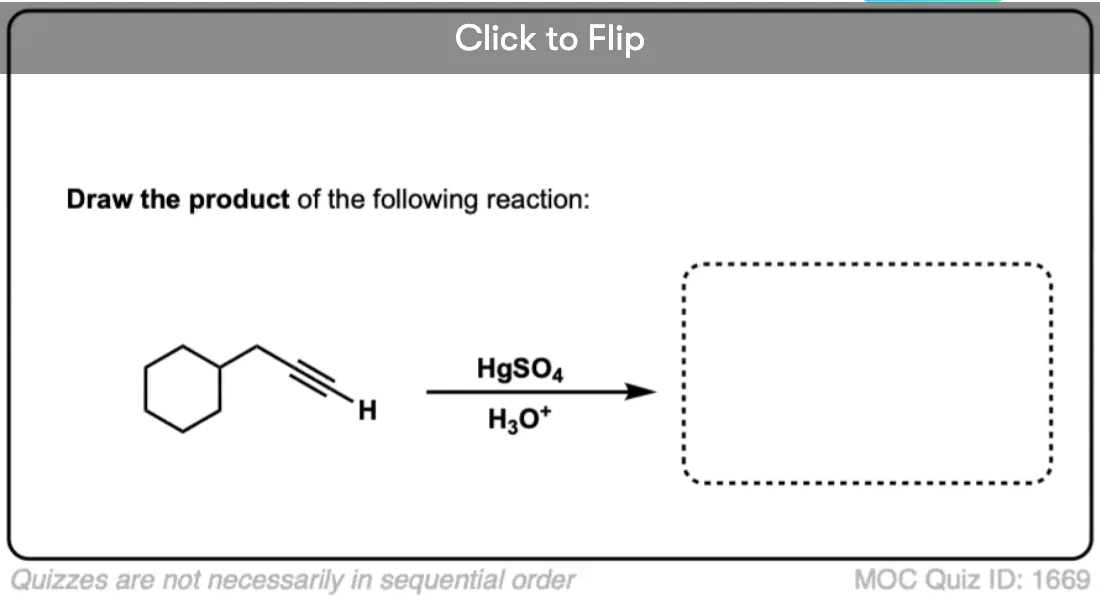
Become a MOC member to see the clickable quiz with answers on the back.
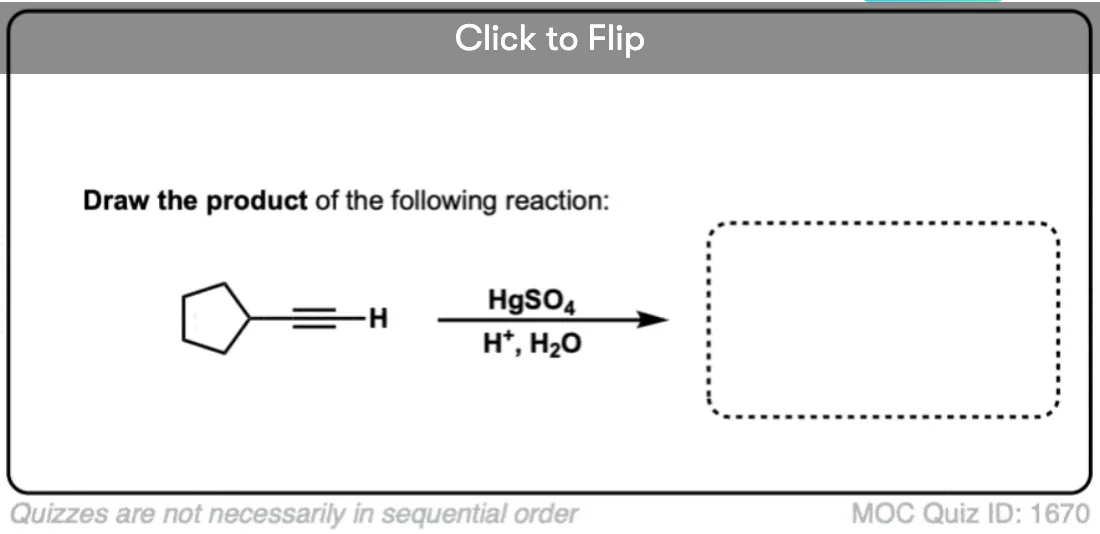
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading
- THE HYDROBORATION OF ACETYLENES – A CONVENIENT CONVERSION OF INTERNAL ACETYLENES TO CIS OLEFINS OF HIGH PURITY AND OF TERMINAL ACETYLENES TO ALDEHYDES
Brown, H.C.; Zweifel, G.
J. Am. Chem. Soc. 1959 81 (6), 1512
DOI: 10.1021/ja01515a058
The original paper describing the hydroboration of alkynes, by Nobel Laureate Prof. H. C. Brown (Purdue). - PALLADIUM-CATALYZED REACTION OF 1-ALKENYLBORONATES WITH VINYLIC HALIDES: (1Z,3E)-1-PHENYL-1,3-OCTADIENE
Miayura, N.; Suzuki, A.
Org. Synth. 1990, 68, 130
DOI:15227/orgsyn.068.0130
A procedure by Nobel Laureate Akira Suzuki for the hydroboration of an alkyne with catecholborane. The resulting product can then be subsequently used in a Pd-catalyzed Suzuki coupling reaction.
A variety of other reagents were developed by H. C. Brown for hydroboration, including catecholborane, 9-BBN, and disiamylborane. The advantage with these reagents is that they will undergo monoadditionto alkynes, whereas borane will add twice. Representative references for the reaction of these reagents with alkynes are below: - Catecholborane (1,3,2-Benzodioxaborole) as a New, General Monohydroboration Reagent for Alkynes. A Convenient Synthesis of Alkeneboronic Esters and Acids from Alkynes via Hydroboration
Brown, H. C.; Gupta, S. K.
J. Am. Chem. Soc. 1972 94(12), 4370
DOI: 10.1021/ja00767a072 - 50. Hydroboration of Representative Alkynes with 9-Borabicyclo[3.3.1]nonane-a Simple Synthesis of Versatile Vinyl Bora and gem-Dibora Intermediates
Brown, H. C.; Scouten, C. G.; Liotta, R.
J. Am. Chem. Soc. 1979 101 (1), 96
DOI:10.1021/ja00495a016 - XI. The Hydroboration of Acetylenes-A Convenient Conversion of Internal Acetylenes into cis-Olefins and of Terminal Acetylenes into Aldehydes
Brown, H. C.; Zweifel, G.
J. Am. Chem. Soc. 1961, 83(18), 3834
DOI: 10.1021/ja01479a024
This paper describes the use of disiamylborane for the selective monohydroboration of alkynes. - UNSATURATION PHENOMENA OF ACETYLENIC ACIDS AND ESTERS. III. THE CONSTITUTION OF SOME MERCURY DERIVATIVES
William Whalley Myddleton, Arthur W. Barrett, and John H. Seager
Journal of the American Chemical Society 1930, 52 (11), 4405-4411
DOI: 10.1021/ja01374a032
One of the earliest reports on oxymercuration in the literature. - 1-ACETYLCYCLOHEXANOL
Gardner W. Stacy and Richard A. Mikulec
Org. Synth. 1955, 35, 1
DOI: 10.15227/orgsyn.035.0001
A pretty standard oxymercuration-hydration reaction of a terminal alkyne in Organic Syntheses, a well-regarded source of independently verified and reproducible organic chemistry laboratory procedures. - Enol acetates, enol ethers, and amines by mercuration of acetylenes
Paul F. Hudrlik and Anne M. Hudrlik
The Journal of Organic Chemistry 1973, 38 (25), 4254-4258
DOI: 10.1021/jo00964a009
The authors in this publication show that the intermediate mercurinum ion can react with nucleophiles other than water, expanding the scope of this reaction. - An efficient synthesis of .gamma.-methylene-.gamma.-butyrolactone (.alpha.’-angelicalactone). Application to the synthesis of deoxyobtusilactone and deoxyisoobtusilactone
Richard A. Amos and John A. Katzenellenbogen
The Journal of Organic Chemistry 1978, 43 (4), 560-564
DOI: 10.1021/jo00398a007
Mercurinium ions can also undergo intramolecular cyclizations as well – in this case, the terminal alkyne can cyclize with the carboxylic acid on the other end in the presence of Hg salts to yield lactones.Prof. Bassetti (Italy) published a nice series of papers on the mechanism of mercurinium ion formation from alkynes: - Metalation of alkynes. 1. Effect of alkyne structure on the rate of acetoxymercuration
Mauro Bassetti and Barbara Floris
The Journal of Organic Chemistry 1986, 51 (22), 4140-4143
DOI: 10.1021/jo00372a007 - Metalation of alkynes. Part 2. Behaviour of alkynes with mercury(II) acetate in methanol: a systematic reinvestigation
Mauro Bassetti and Barbara Floris
J. Chem. Soc., Perkin Trans. 2, 1988, 227-233
DOI: 10.1039/P29880000227 - Geminal Organometallic Compounds. I. The Synthesis and Structure of 1,1-Diborohexane
G. Zweifel and H. Arzoumanian
Journal of the American Chemical Society 1967 89 (2), 291-295
DOI: 10.1021/ja00978a022
1-Hexyne undergoes double hydroboration reaction when treated with BH3. This is why bulkier hydroboration reagents such as disiamyl borane or 9-BBN are used.
I was trying to figure out what the mechanism of Oxymercuration of alkynes might look like and I have a doubt…
Do the same steps of Hydration of Alkynes i.e.
1. Formation of Enol and 2. Tautomerization, occur during Oxymercuration rxns?
By definition a chiral center must have four different groups attached. Two hydrogens are delivered to the alpha carbon of the ketone. Therefore formation of enantiomers is not possible in this reaction.
In part 4(mechanism of addition of water),in step2(tautomerization)
i want to ask about hydrogen that h3O donates.can that H be added by any side so we will have enantiomers or it is stereospecific?
What an amazing page ! It cleared all my doubts regarding this topic . My Chemistry teacher couldn’t have taught it better :) Please keep up the good work .
If water are added via oxymercurium path, don’t a second stage, of using hydroboran to reduce oxymercurium inermidiate, is needed?
I know that in the case of alkenes and these reactions (both with Hg and BH3) rearrangements are not observed.
Is rearrangement possible here? (as to form a more substitued thus more stable C+)
Thanks in advance.
No, there’s no free carbocation.
In the very last example (“One final note”), why don’t we use carbocation stability, the reason for Markovnikov’s Rule?
We could say that the Carbon on the right would form a more stable carbocation, because of hyperconjugation. (As it has 3 alpha-hydrogens while the left carbon has only 2)
So this would mean that the 2nd product would dominate.
Or am I missing something?
If an alkyne was on the end of a molecule and being reacted, how would it react with H2SO4 and HgSO4. Would it be an aldehyde or a ketone?
For H2so4 reactions its always a ketone for Bh3 reaction it can be ketone or aldehyde
Hydration of a terminal alkyne will first form an enol following Markovnikov’s rule. The enol will then undergo Keto-Enol Tautomerization and rearrange to a methyl ketone.
it will be a ketone
In the mechanism “Step 1 – enol formation” a plus charge is missing in the 3rd structure.
fixed. Thank you!
What I would really like to know is that WHY THE ANTIMARKONIKOV product is formed in the Hydroboration reactions.Basically I want the simplified mechanism which I can’t find in my books.
And congratulations of becoming a Dad, Mr.James.
It has to do with the fact that hydrogen is more electronegative than boron.
Is the regioselectivity of BH3 any good? Seems like hydroboration via 9-BBN or some other bulky R2BH reagent greatly enhances the anti-Markovnikov product via sterics. While the electronegativity difference is real, you’re hoping for a collision that leads to the π bond attacking boron for the syn transfer of the H to the more substituted side of the alkene (or alkyne)–is there any way something as small as BH3 can reliably select for the anti-M product?
Thanks for this comment. The problem is not regioselectivity but that 1,1-dibora products form, which is why bulky boranes must be used. See JACS 1967 vol. 89 p. 291. https://pubs.acs.org/doi/10.1021/ja00978a022
You made this so easy to understand. thanks! :)